

Protein target highlights in CASP15: Analysis of models by structure providers
- PMID: 37493353
- PMCID: PMC10792529
- DOI: 10.1002/prot.26545
Protein target highlights in CASP15: Analysis of models by structure providers
Abstract
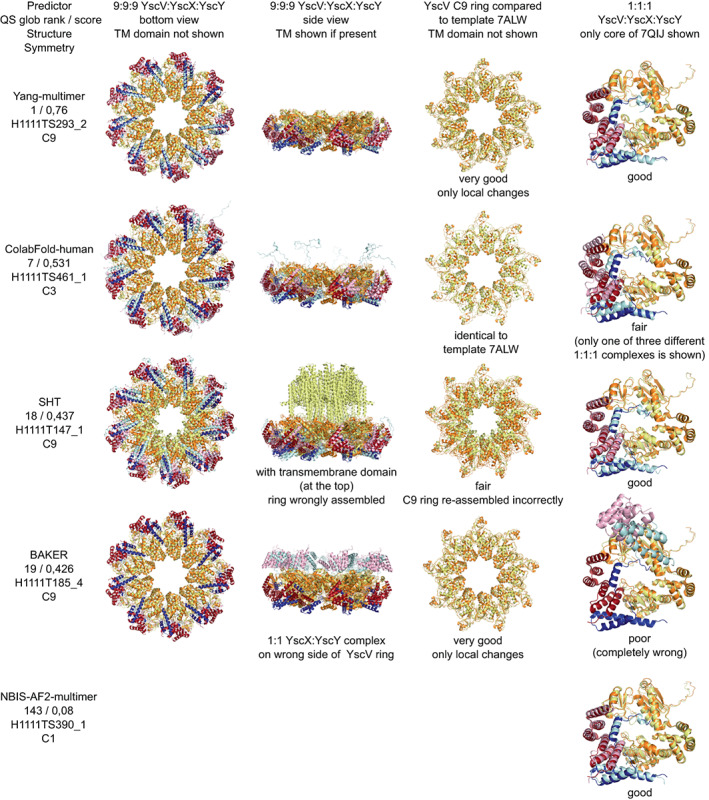
We present an in-depth analysis of selected CASP15 targets, focusing on their biological and functional significance. The authors of the structures identify and discuss key protein features and evaluate how effectively these aspects were captured in the submitted predictions. While the overall ability to predict three-dimensional protein structures continues to impress, reproducing uncommon features not previously observed in experimental structures is still a challenge. Furthermore, instances with conformational flexibility and large multimeric complexes highlight the need for novel scoring strategies to better emphasize biologically relevant structural regions. Looking ahead, closer integration of computational and experimental techniques will play a key role in determining the next challenges to be unraveled in the field of structural molecular biology.
Keywords: CASP; X-ray crystallography; cryo-EM; protein structure prediction.
© 2023 The Authors. Proteins: Structure, Function, and Bioinformatics published by Wiley Periodicals LLC.
Figures















References
-
- Kryshtafovych A, Moult J, Bales P, et al. Challenging the state of the art in protein structure prediction: highlights of experimental target structures for the 10th critical assessment of techniques for protein structure prediction experiment CASP10. Proteins. 2014;82(Suppl 2):26‐42. - PMC - PubMed
MeSH terms
Substances
Grants and funding
- R01 CA217255/CA/NCI NIH HHS/United States
- 75N93022C00035/AI/NIAID NIH HHS/United States
- R35 GM133706/GM/NIGMS NIH HHS/United States
- S10 OD018111/OD/NIH HHS/United States
- R01 GM100482/GM/NIGMS NIH HHS/United States
- P20 GM121176/GM/NIGMS NIH HHS/United States
- R35 GM128777/GM/NIGMS NIH HHS/United States
- R21 CA255894/CA/NCI NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
- R01 AI134678/AI/NIAID NIH HHS/United States
- R01 AI174646/AI/NIAID NIH HHS/United States
- R01 GM115261/GM/NIGMS NIH HHS/United States
- R01 GM139978/GM/NIGMS NIH HHS/United States
- R01 GM071940/GM/NIGMS NIH HHS/United States
- R01 AI165079/AI/NIAID NIH HHS/United States
- S10 RR023057/RR/NCRR NIH HHS/United States
