

Tutorial: a statistical genetics guide to identifying HLA alleles driving complex disease
- PMID: 37495751
- PMCID: PMC10786448
- DOI: 10.1038/s41596-023-00853-4
Tutorial: a statistical genetics guide to identifying HLA alleles driving complex disease
Abstract
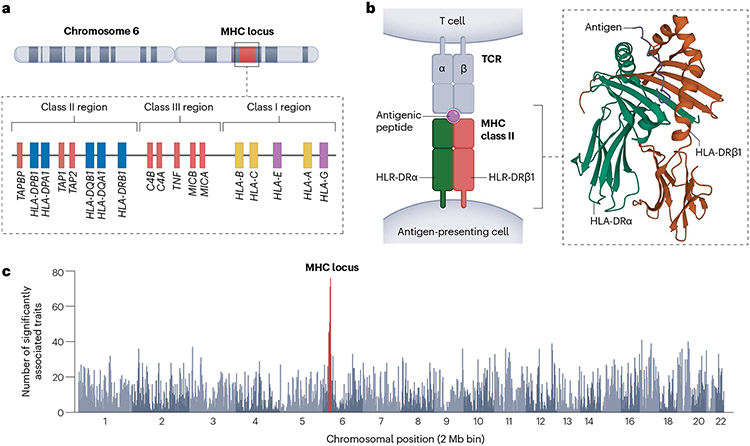
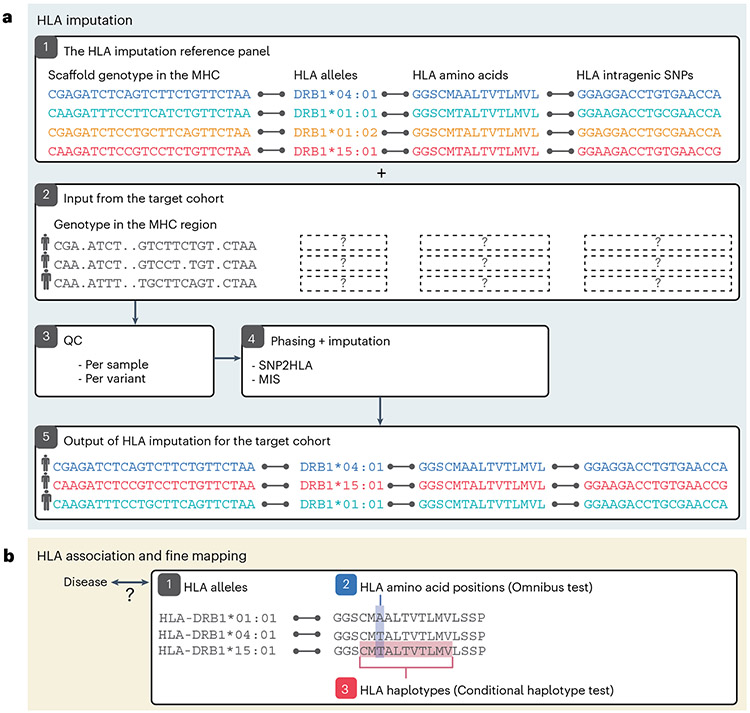
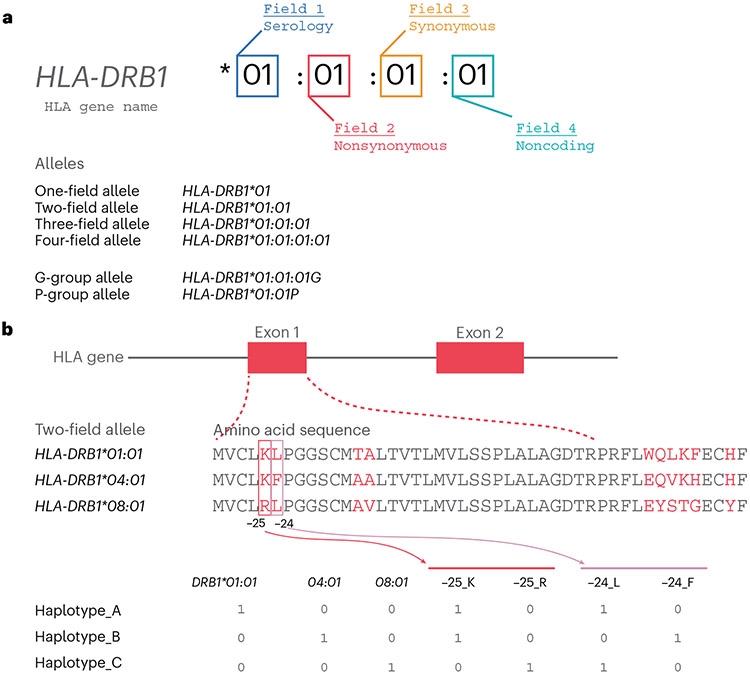
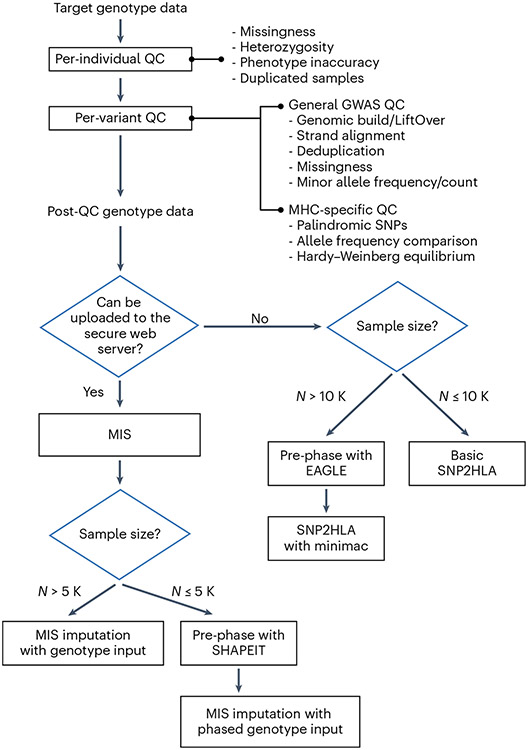
The human leukocyte antigen (HLA) locus is associated with more complex diseases than any other locus in the human genome. In many diseases, HLA explains more heritability than all other known loci combined. In silico HLA imputation methods enable rapid and accurate estimation of HLA alleles in the millions of individuals that are already genotyped on microarrays. HLA imputation has been used to define causal variation in autoimmune diseases, such as type I diabetes, and in human immunodeficiency virus infection control. However, there are few guidelines on performing HLA imputation, association testing, and fine mapping. Here, we present a comprehensive tutorial to impute HLA alleles from genotype data. We provide detailed guidance on performing standard quality control measures for input genotyping data and describe options to impute HLA alleles and amino acids either locally or using the web-based Michigan Imputation Server, which hosts a multi-ancestry HLA imputation reference panel. We also offer best practice recommendations to conduct association tests to define the alleles, amino acids, and haplotypes that affect human traits. Along with the pipeline, we provide a step-by-step online guide with scripts and available software ( https://github.com/immunogenomics/HLA_analyses_tutorial ). This tutorial will be broadly applicable to large-scale genotyping data and will contribute to defining the role of HLA in human diseases across global populations.
© 2023. Springer Nature Limited.
Figures
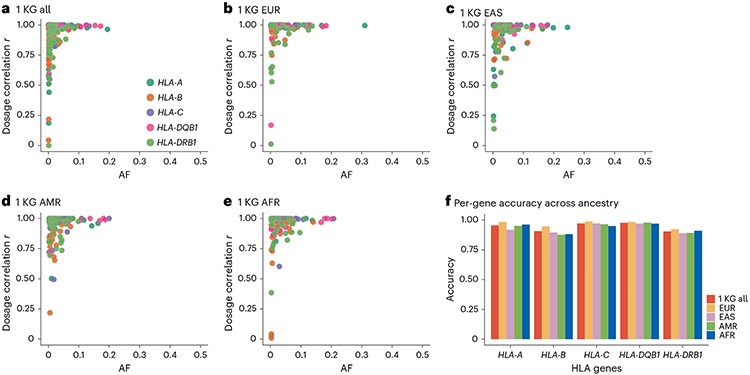

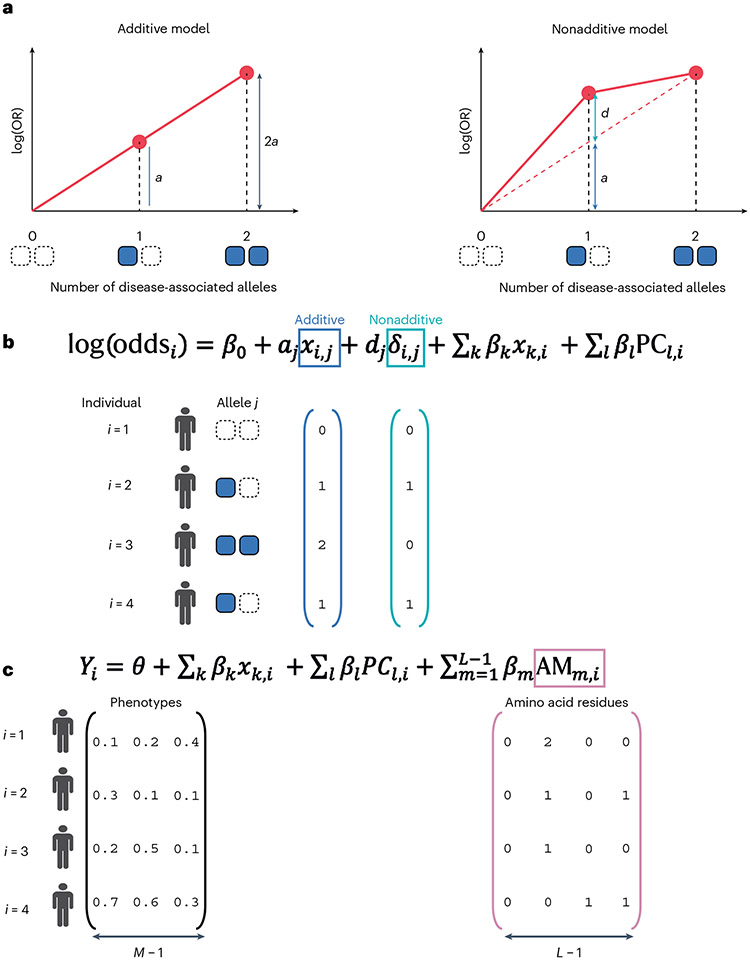
References
-
- Amiel J. in Histocompatibility Testing (ed. Teraski PI) 79–81 (Munksgaard, 1967).
-
- Murphy K & Weaver C Janeway’s immunology. America 1–277 (2017).
-
- Dendrou CA, Petersen J, Rossjohn J & Fugger L HLA variation and disease. Nat. Rev. Immunol 18, 325–339 (2018). - PubMed
-
- Murphy K Kenneth M & Weaver C Janeway’s Immunobiology (Garland Science, 2016).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
