

Darier's disease exhibits a unique cutaneous microbial dysbiosis associated with inflammation and body malodour
- PMID: 37496039
- PMCID: PMC10369845
- DOI: 10.1186/s40168-023-01587-x
Darier's disease exhibits a unique cutaneous microbial dysbiosis associated with inflammation and body malodour
Erratum in
-
Correction: Darier's disease exhibits a unique cutaneous microbial dysbiosis associated with inflammation and body malodour.Microbiome. 2023 Sep 14;11(1):206. doi: 10.1186/s40168-023-01665-0. Microbiome. 2023. PMID: 37705089 Free PMC article. No abstract available.
Abstract
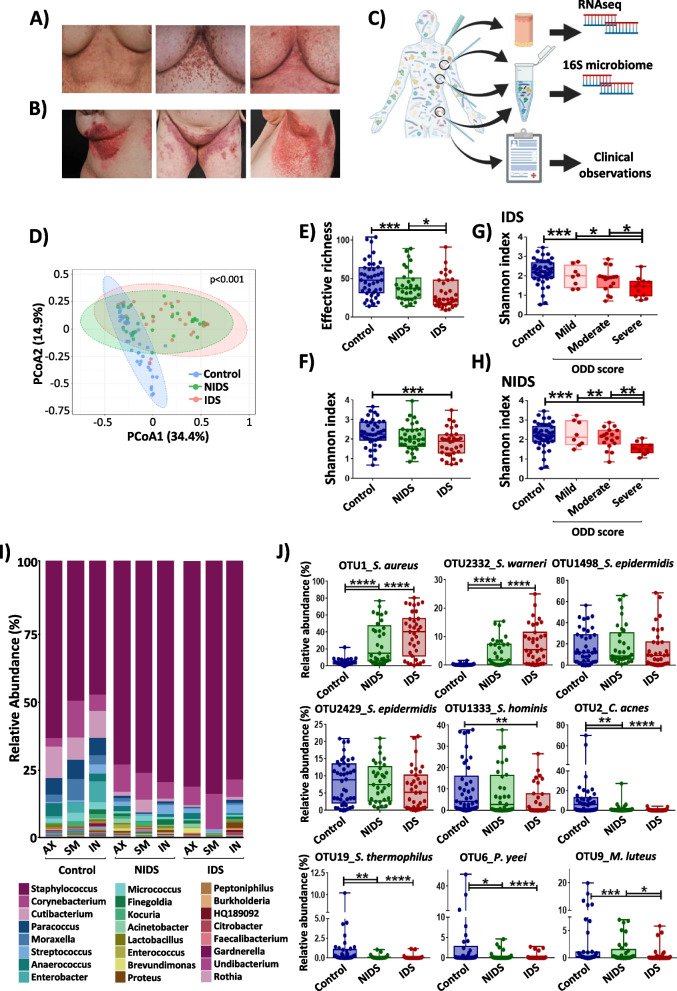
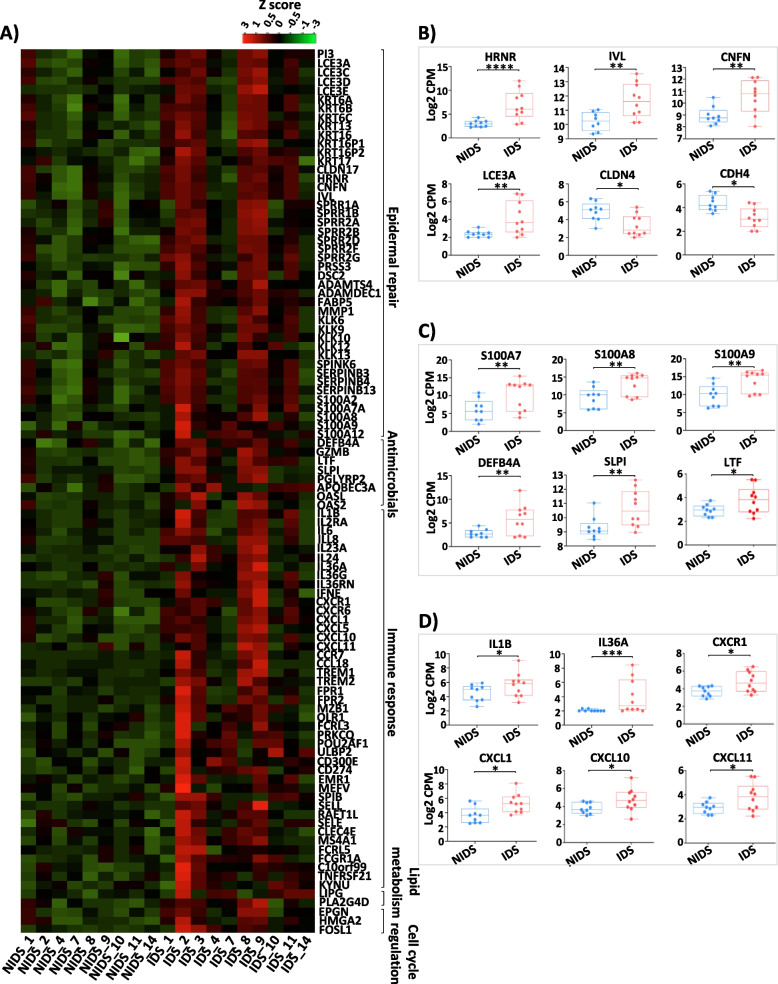
Background: Darier's disease (DD) is a genodermatosis caused by mutations of the ATP2A2 gene leading to disrupted keratinocyte adhesion. Recurrent episodes of skin inflammation and infections with a typical malodour in DD indicate a role for microbial dysbiosis. Here, for the first time, we investigated the DD skin microbiome using a metabarcoding approach of 115 skin swabs from 14 patients and 14 healthy volunteers. Furthermore, we analyzed its changes in the context of DD malodour and the cutaneous DD transcriptome.
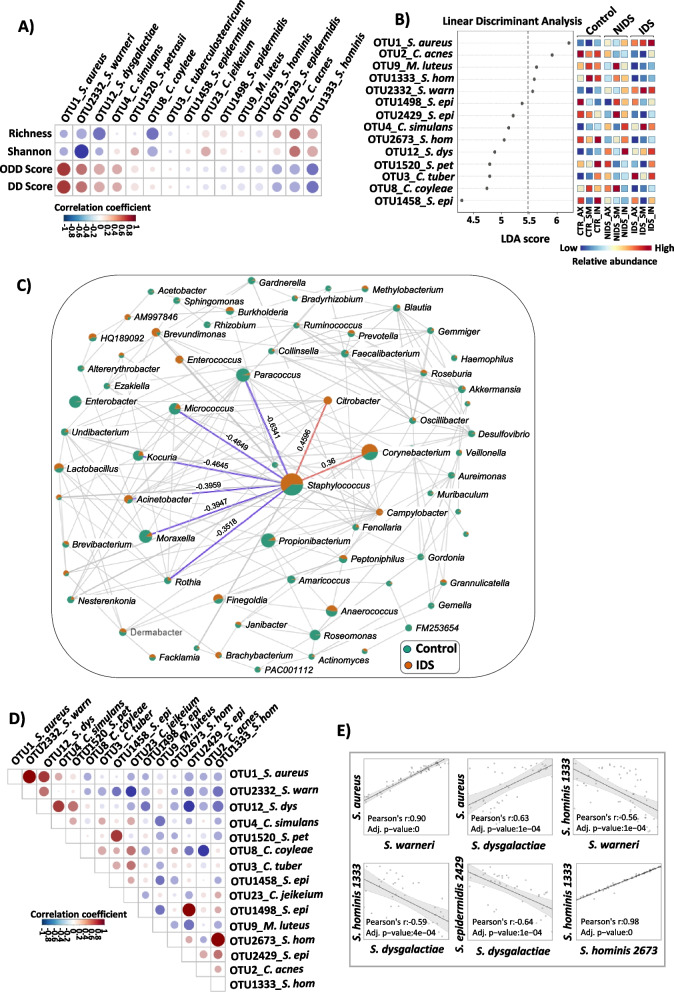
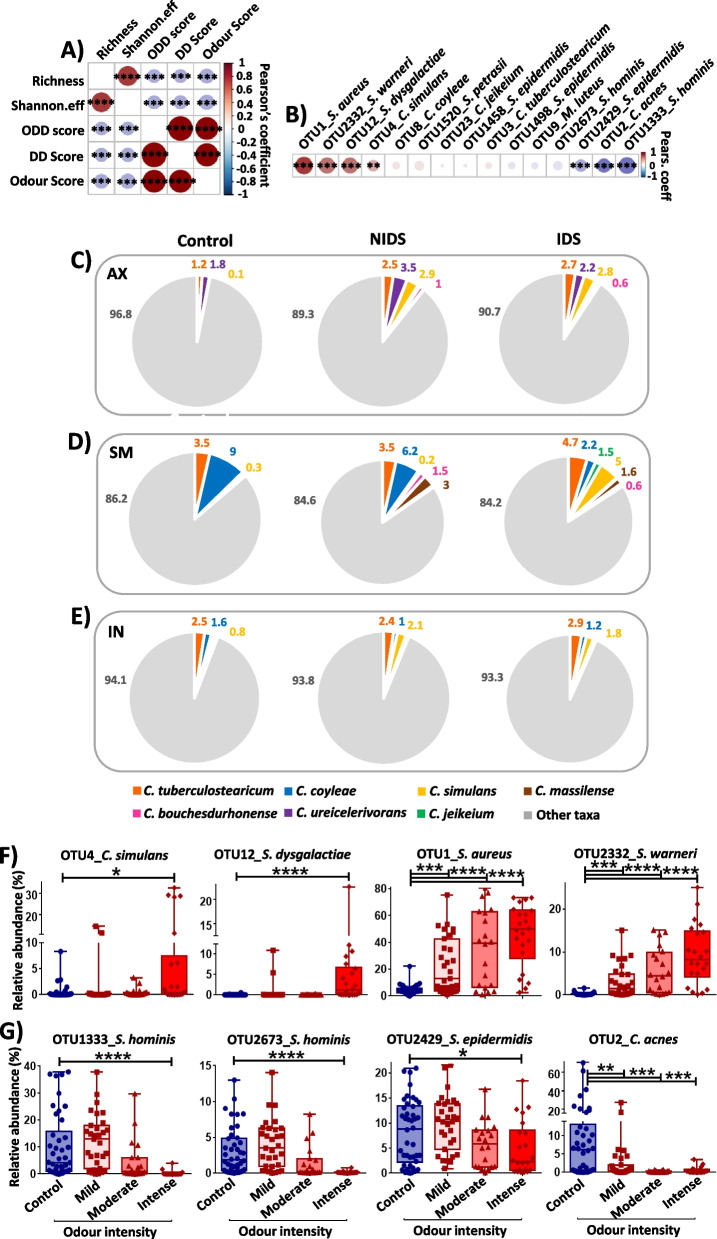
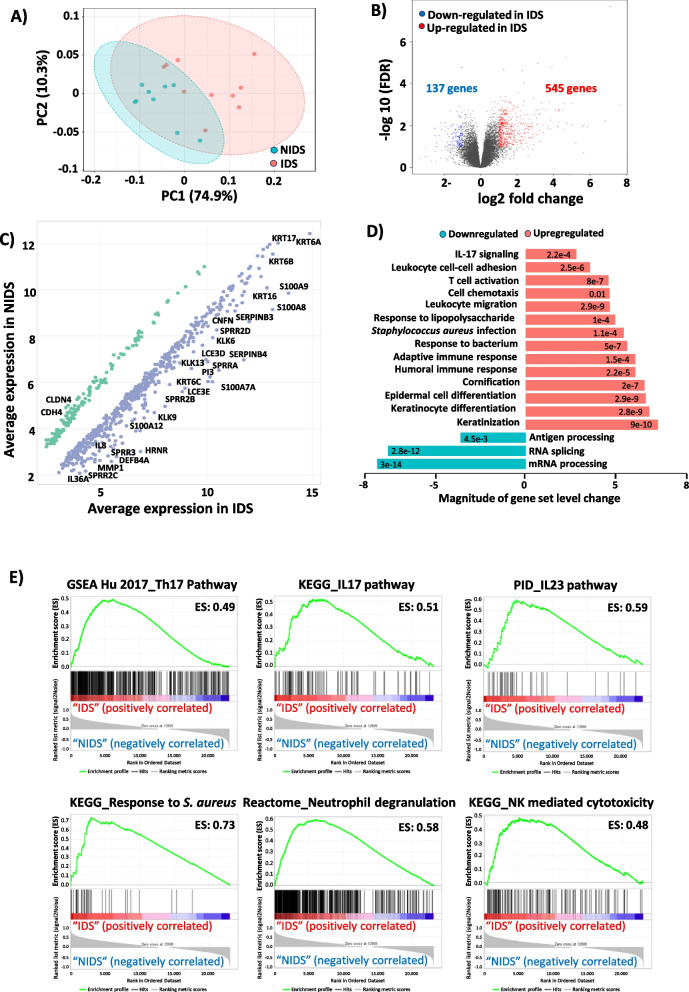
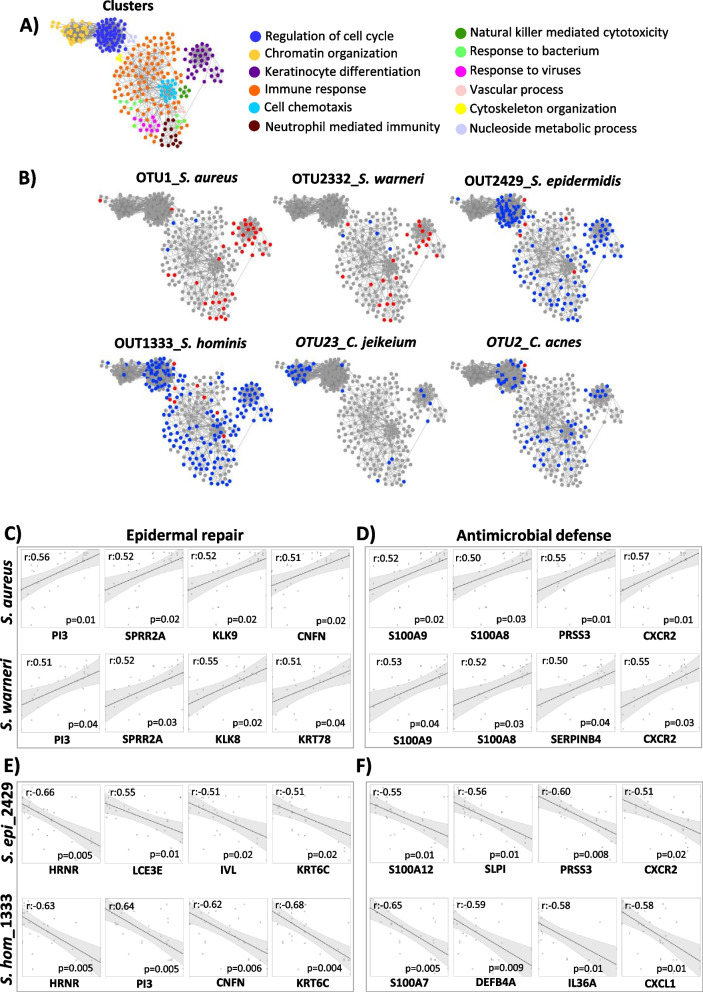
Results: We identified a disease-specific cutaneous microbiome with a loss of microbial diversity and of potentially beneficial commensals. Expansion of inflammation-associated microbes such as Staphylococcus aureus and Staphylococcus warneri strongly correlated with disease severity. DD dysbiosis was further characterized by abundant species belonging to Corynebacteria, Staphylococci and Streptococci groups displaying strong associations with malodour intensity. Transcriptome analyses showed marked upregulation of epidermal repair, inflammatory and immune defence pathways reflecting epithelial and immune response mechanisms to DD dysbiotic microbiome. In contrast, barrier genes including claudin-4 and cadherin-4 were downregulated.
Conclusions: These findings allow a better understanding of Darier exacerbations, highlighting the role of cutaneous dysbiosis in DD inflammation and associated malodour. Our data also suggest potential biomarkers and targets of intervention for DD. Video Abstract.
Keywords: Darier’s disease; Genodermatosis; Malodour; Microbiome; Skin barrier; Transcriptome.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Clinical case of acral hemorrhagic Darier's disease is not caused by mutations in exon 15 of the ATP2A2 gene.Coll Antropol. 2003 Jun;27(1):125-33. Coll Antropol. 2003. PMID: 12974140
-
Five mutations of ATP2A2 gene in Chinese patients with Darier's disease and a literature review of 86 cases reported in China.Arch Dermatol Res. 2006 Jul;298(2):58-63. doi: 10.1007/s00403-006-0658-0. Epub 2006 Mar 22. Arch Dermatol Res. 2006. PMID: 16552539 Review.
-
Seven novel mutations in the ATP2A2 gene of Austrian patients with Darier's disease.Arch Dermatol Res. 2011 Jul;303(5):371-4. doi: 10.1007/s00403-011-1148-6. Epub 2011 Apr 26. Arch Dermatol Res. 2011. PMID: 21519848
-
Exacerbation, then clearance, of mutation-proven Darier's disease of the skin after radiotherapy for bronchial carcinoma: a case of radiation-induced epidermal differentiation?Radiat Res. 2001 Dec;156(6):724-30. doi: 10.1667/0033-7587(2001)156[0724:etcomp]2.0.co;2. Radiat Res. 2001. PMID: 11741496
-
Mendelian Disorders of Cornification Caused by Defects in Intracellular Calcium Pumps: Mutation Update and Database for Variants in ATP2A2 and ATP2C1 Associated with Darier Disease and Hailey-Hailey Disease.Hum Mutat. 2017 Apr;38(4):343-356. doi: 10.1002/humu.23164. Epub 2017 Feb 15. Hum Mutat. 2017. PMID: 28035777 Review.
Cited by
-
Correction: Darier's disease exhibits a unique cutaneous microbial dysbiosis associated with inflammation and body malodour.Microbiome. 2023 Sep 14;11(1):206. doi: 10.1186/s40168-023-01665-0. Microbiome. 2023. PMID: 37705089 Free PMC article. No abstract available.
-
Darier disease: Current insights and challenges in pathogenesis and management.J Eur Acad Dermatol Venereol. 2025 May;39(5):942-951. doi: 10.1111/jdv.20448. Epub 2024 Nov 28. J Eur Acad Dermatol Venereol. 2025. PMID: 39606894 Free PMC article. Review.
-
Th17-associated cytokines IL-17 and IL-23 in inflamed skin of Darier disease patients as potential therapeutic targets.Nat Commun. 2023 Nov 17;14(1):7470. doi: 10.1038/s41467-023-43210-5. Nat Commun. 2023. PMID: 37978298 Free PMC article.
-
Pruritus and Neuropsychiatric Symptoms Among Patients with Darier Disease-An Overlooked and Interconnected Challenge.J Clin Med. 2025 Mar 8;14(6):1818. doi: 10.3390/jcm14061818. J Clin Med. 2025. PMID: 40142630 Free PMC article.
-
Exploring the Risk of Alcohol Addiction and Substance Use Disorders in Darier Disease and Hailey-Hailey Disease.Acta Derm Venereol. 2025 Jun 25;105:adv43756. doi: 10.2340/actadv.v105.43756. Acta Derm Venereol. 2025. PMID: 40557968 Free PMC article. No abstract available.
References
-
- Darier J. Psorospermose folliculaire végétante. Ann Dermatol Syphiligr. 1889;10:597–612.
-
- White JC. A case of keratosis (icthyosis) follicularis. Cutan Genitourin Dis. 1889;7:201–209.
-
- Svendsen IB, Albrectsen B. The prevalence of dyskeratosis follicularis (Darier’s disease) in Denmark: an investigation of the heredity in 22 families. Acta Derm Venereol. 1959;39:256–269. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
