

Online tree expansion could help solve the problem of scalability in Bayesian phylogenetics
- PMID: 37498209
- PMCID: PMC10627553
- DOI: 10.1093/sysbio/syad045
Online tree expansion could help solve the problem of scalability in Bayesian phylogenetics
Abstract
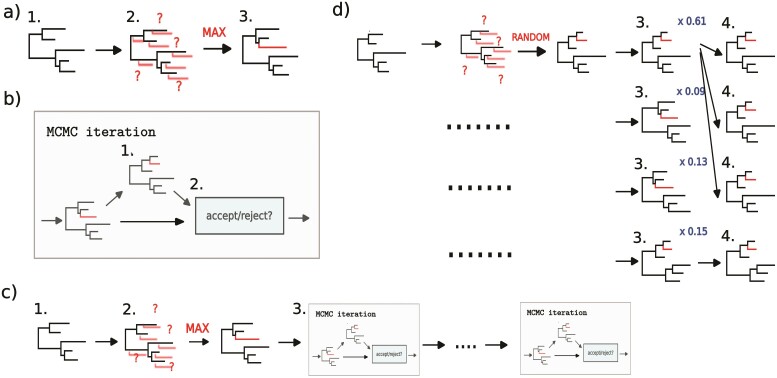
Bayesian phylogenetics is now facing a critical point. Over the last 20 years, Bayesian methods have reshaped phylogenetic inference and gained widespread popularity due to their high accuracy, the ability to quantify the uncertainty of inferences and the possibility of accommodating multiple aspects of evolutionary processes in the models that are used. Unfortunately, Bayesian methods are computationally expensive, and typical applications involve at most a few hundred sequences. This is problematic in the age of rapidly expanding genomic data and increasing scope of evolutionary analyses, forcing researchers to resort to less accurate but faster methods, such as maximum parsimony and maximum likelihood. Does this spell doom for Bayesian methods? Not necessarily. Here, we discuss some recently proposed approaches that could help scale up Bayesian analyses of evolutionary problems considerably. We focus on two particular aspects: online phylogenetics, where new data sequences are added to existing analyses, and alternatives to Markov chain Monte Carlo (MCMC) for scalable Bayesian inference. We identify 5 specific challenges and discuss how they might be overcome. We believe that online phylogenetic approaches and Sequential Monte Carlo hold great promise and could potentially speed up tree inference by orders of magnitude. We call for collaborative efforts to speed up the development of methods for real-time tree expansion through online phylogenetics.
Keywords: Bayesian inference; MCMC; phylogeny; sequential Monte Carlo.
© The Author(s) 2023. Published by Oxford University Press on behalf of the Society of Systematic Biologists.
Figures
Similar articles
-
Phylogenetic inference via sequential Monte Carlo.Syst Biol. 2012 Jul;61(4):579-93. doi: 10.1093/sysbio/syr131. Epub 2012 Jan 4. Syst Biol. 2012. PMID: 22223445 Free PMC article.
-
Identifiability of parameters in MCMC Bayesian inference of phylogeny.Syst Biol. 2002 Oct;51(5):754-60. doi: 10.1080/10635150290102429. Syst Biol. 2002. PMID: 12396589
-
Scalable Bayesian phylogenetics.Philos Trans R Soc Lond B Biol Sci. 2022 Oct 10;377(1861):20210242. doi: 10.1098/rstb.2021.0242. Epub 2022 Aug 22. Philos Trans R Soc Lond B Biol Sci. 2022. PMID: 35989603 Free PMC article. Review.
-
An Annealed Sequential Monte Carlo Method for Bayesian Phylogenetics.Syst Biol. 2020 Jan 1;69(1):155-183. doi: 10.1093/sysbio/syz028. Syst Biol. 2020. PMID: 31173141
-
A biologist's guide to Bayesian phylogenetic analysis.Nat Ecol Evol. 2017 Oct;1(10):1446-1454. doi: 10.1038/s41559-017-0280-x. Epub 2017 Sep 21. Nat Ecol Evol. 2017. PMID: 28983516 Free PMC article. Review.
Cited by
-
Modeling Substitution Rate Evolution across Lineages and Relaxing the Molecular Clock.Genome Biol Evol. 2024 Sep 3;16(9):evae199. doi: 10.1093/gbe/evae199. Genome Biol Evol. 2024. PMID: 39332907 Free PMC article. Review.
-
Challenges in Assembling the Dated Tree of Life.Genome Biol Evol. 2024 Oct 9;16(10):evae229. doi: 10.1093/gbe/evae229. Genome Biol Evol. 2024. PMID: 39475308 Free PMC article.
-
Phylogenetic Tree Instability After Taxon Addition: Empirical Frequency, Predictability, and Consequences For Online Inference.Syst Biol. 2025 Feb 10;74(1):101-111. doi: 10.1093/sysbio/syae059. Syst Biol. 2025. PMID: 39453463 Free PMC article.
References
-
- Andrieu C., Doucet A., Holenstein R.. 2010. Particle Markov chain Monte Carlo methods. J.R. Stat. Soc. 72(3):269–342
-
- Atteson K. 1999. The performance of neighbor-joining methods of phylogenetic reconstruction. Algorithmica. 25(2):251–278.
-
- Ayres D.L., Cummings M.P., Baele G., Darling A.E., Lewis P.O., Swofford D.L., Huelsenbeck J.P., Lemey P., Rambaut A., Suchard M.A.. 2019. Beagle 3: improved performance, scaling, and usability for a high-performance computing library for statistical phylogenetics. Syst. Biol. 68(6):1052–1061. - PMC - PubMed
-
- Balaban M., Jiang Y., Roush D., Zhu Q., Mirarab S.. 2022. Fast and accurate distance-based phylogenetic placement using divide and conquer. Mol. Ecol. Resour. 22(3):1213–1227. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
