

Deploying synthetic coevolution and machine learning to engineer protein-protein interactions
- PMID: 37499032
- PMCID: PMC10403280
- DOI: 10.1126/science.adh1720
Deploying synthetic coevolution and machine learning to engineer protein-protein interactions
Abstract
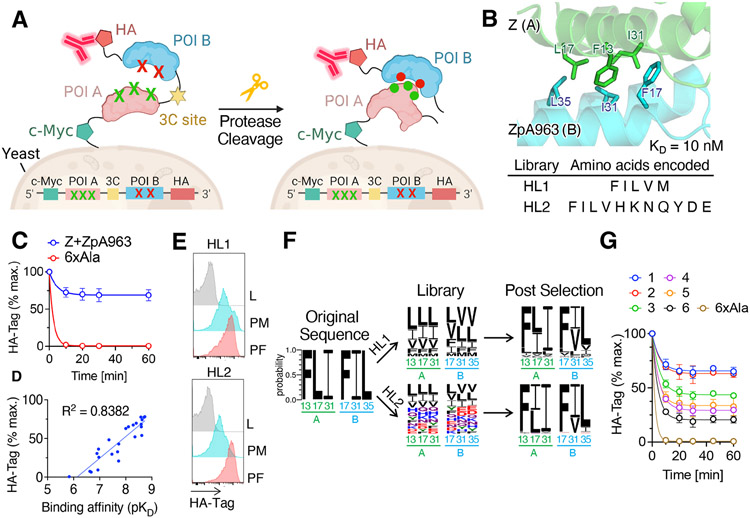
Fine-tuning of protein-protein interactions occurs naturally through coevolution, but this process is difficult to recapitulate in the laboratory. We describe a platform for synthetic protein-protein coevolution that can isolate matched pairs of interacting muteins from complex libraries. This large dataset of coevolved complexes drove a systems-level analysis of molecular recognition between Z domain-affibody pairs spanning a wide range of structures, affinities, cross-reactivities, and orthogonalities, and captured a broad spectrum of coevolutionary networks. Furthermore, we harnessed pretrained protein language models to expand, in silico, the amino acid diversity of our coevolution screen, predicting remodeled interfaces beyond the reach of the experimental library. The integration of these approaches provides a means of simulating protein coevolution and generating protein complexes with diverse molecular recognition properties for biotechnology and synthetic biology.
Figures





References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
