

MSBooster: improving peptide identification rates using deep learning-based features
- PMID: 37500632
- PMCID: PMC10374903
- DOI: 10.1038/s41467-023-40129-9
MSBooster: improving peptide identification rates using deep learning-based features
Abstract
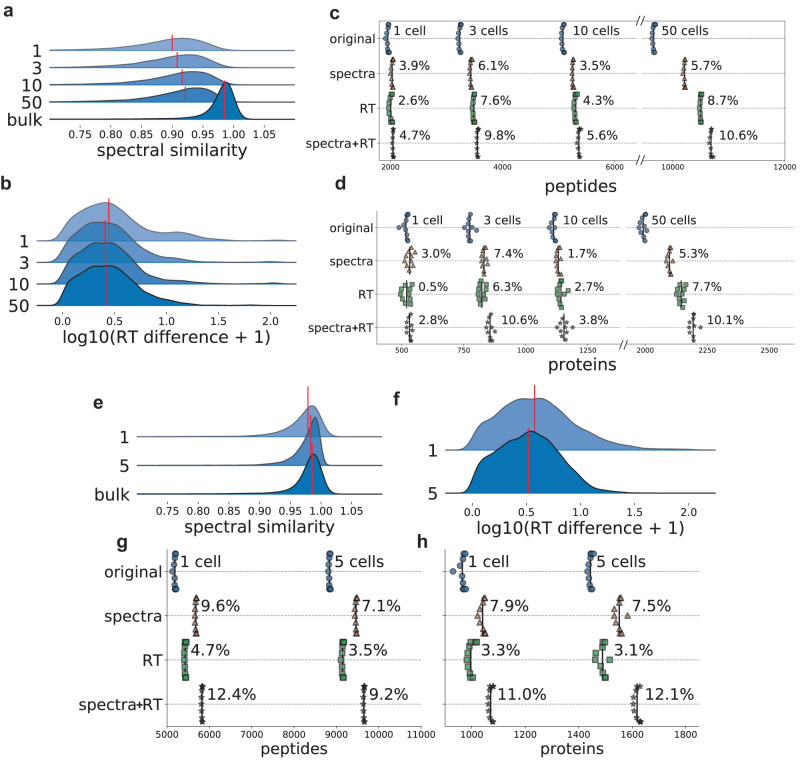
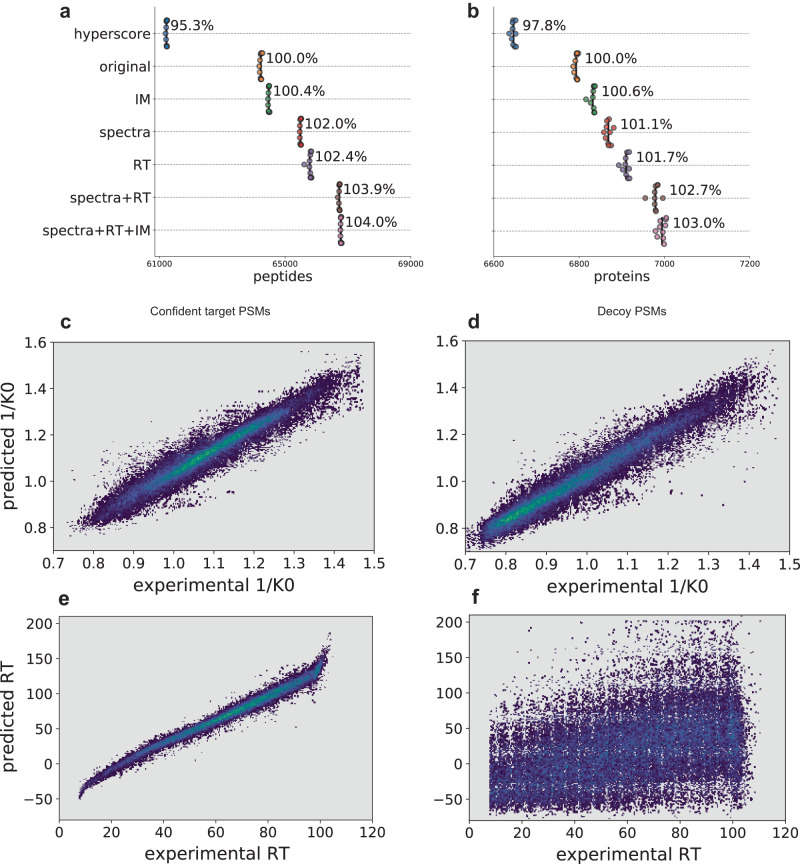
Peptide identification in liquid chromatography-tandem mass spectrometry (LC-MS/MS) experiments relies on computational algorithms for matching acquired MS/MS spectra against sequences of candidate peptides using database search tools, such as MSFragger. Here, we present a new tool, MSBooster, for rescoring peptide-to-spectrum matches using additional features incorporating deep learning-based predictions of peptide properties, such as LC retention time, ion mobility, and MS/MS spectra. We demonstrate the utility of MSBooster, in tandem with MSFragger and Percolator, in several different workflows, including nonspecific searches (immunopeptidomics), direct identification of peptides from data independent acquisition data, single-cell proteomics, and data generated on an ion mobility separation-enabled timsTOF MS platform. MSBooster is fast, robust, and fully integrated into the widely used FragPipe computational platform.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
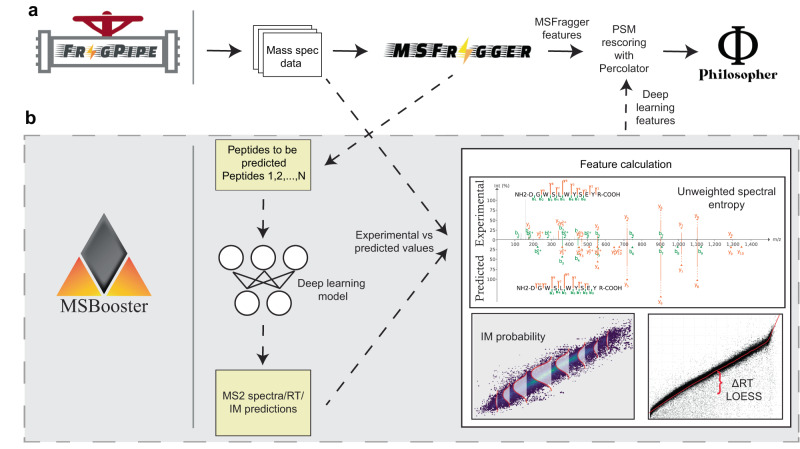
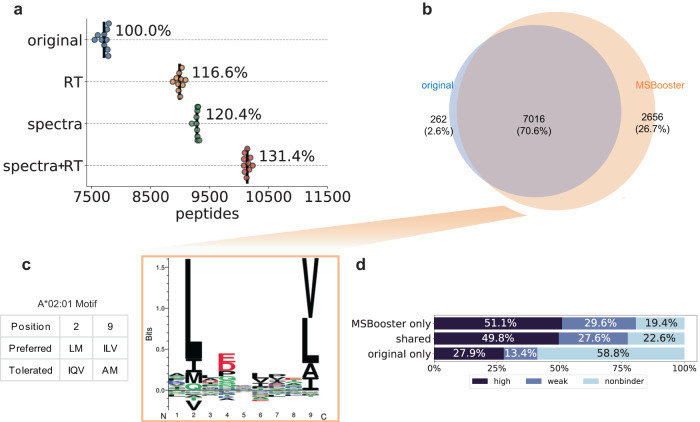
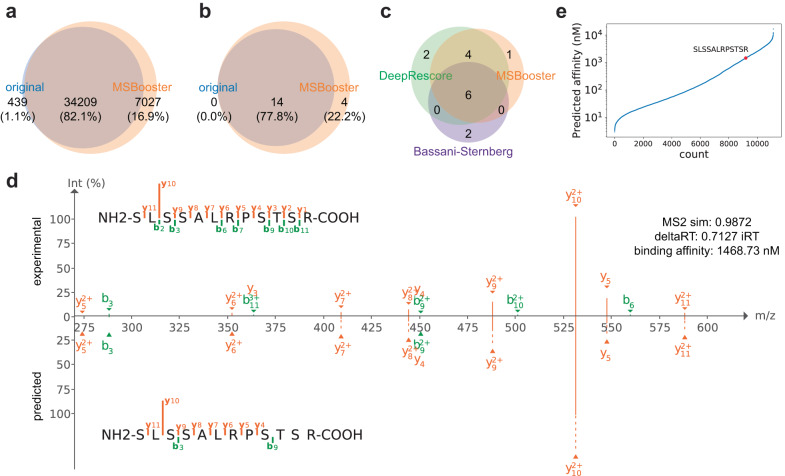
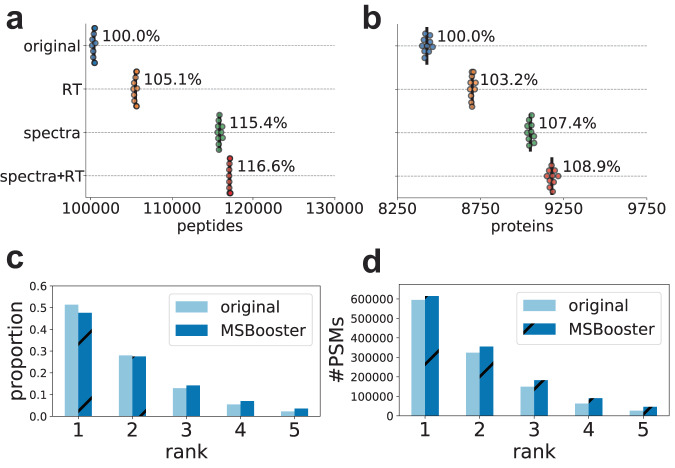
References
-
- Kitata, R. B., Yang, J. C. & Chen, Y. J. Advances in data-independent acquisition mass spectrometry towards comprehensive digital proteome landscape. Mass Spectrom. Rev. e21781 (2022). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
