

Heterozygous rare variants in NR2F2 cause a recognizable multiple congenital anomaly syndrome with developmental delays
- PMID: 37500725
- PMCID: PMC10545729
- DOI: 10.1038/s41431-023-01434-5
Heterozygous rare variants in NR2F2 cause a recognizable multiple congenital anomaly syndrome with developmental delays
Abstract
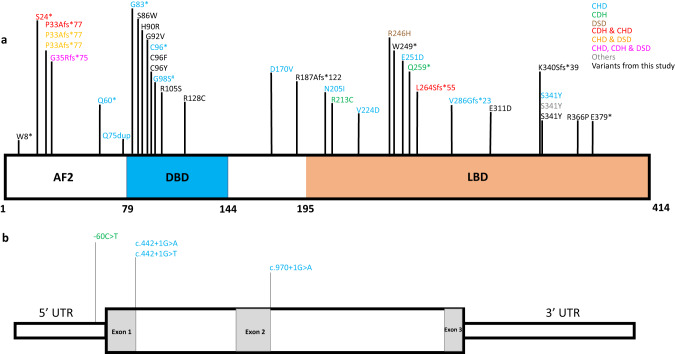
Nuclear receptor subfamily 2 group F member 2 (NR2F2 or COUP-TF2) encodes a transcription factor which is expressed at high levels during mammalian development. Rare heterozygous Mendelian variants in NR2F2 were initially identified in individuals with congenital heart disease (CHD), then subsequently in cohorts of congenital diaphragmatic hernia (CDH) and 46,XX ovotesticular disorders/differences of sexual development (DSD); however, the phenotypic spectrum associated with pathogenic variants in NR2F2 remains poorly characterized. Currently, less than 40 individuals with heterozygous pathogenic variants in NR2F2 have been reported. Here, we review the clinical and molecular details of 17 previously unreported individuals with rare heterozygous NR2F2 variants, the majority of which were de novo. Clinical features were variable, including intrauterine growth restriction (IUGR), CHD, CDH, genital anomalies, DSD, developmental delays, hypotonia, feeding difficulties, failure to thrive, congenital and acquired microcephaly, dysmorphic facial features, renal failure, hearing loss, strabismus, asplenia, and vascular malformations, thus expanding the phenotypic spectrum associated with NR2F2 variants. The variants seen were predicted loss of function, including a nonsense variant inherited from a mildly affected mosaic mother, missense and a large deletion including the NR2F2 gene. Our study presents evidence for rare, heterozygous NR2F2 variants causing a highly variable syndrome of congenital anomalies, commonly associated with heart defects, developmental delays/intellectual disability, dysmorphic features, feeding difficulties, hypotonia, and genital anomalies. Based on the new and previous cases, we provide clinical recommendations for evaluating individuals diagnosed with an NR2F2-associated disorder.
© 2023. The Author(s), under exclusive licence to European Society of Human Genetics.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
