

Integrated single-cell chromatin and transcriptomic analyses of human scalp identify gene-regulatory programs and critical cell types for hair and skin diseases
- PMID: 37500727
- PMCID: PMC11190942
- DOI: 10.1038/s41588-023-01445-4
Integrated single-cell chromatin and transcriptomic analyses of human scalp identify gene-regulatory programs and critical cell types for hair and skin diseases
Abstract
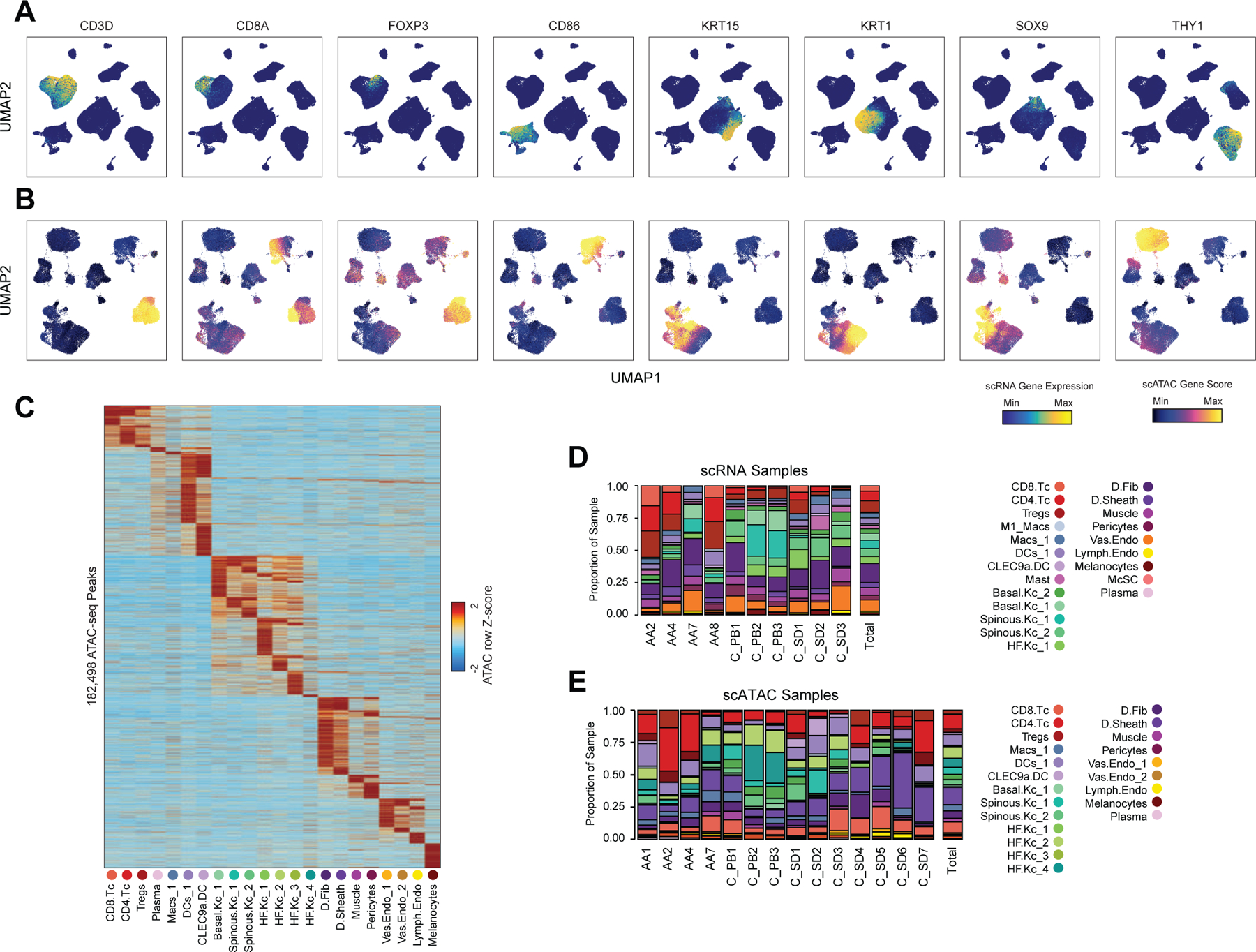
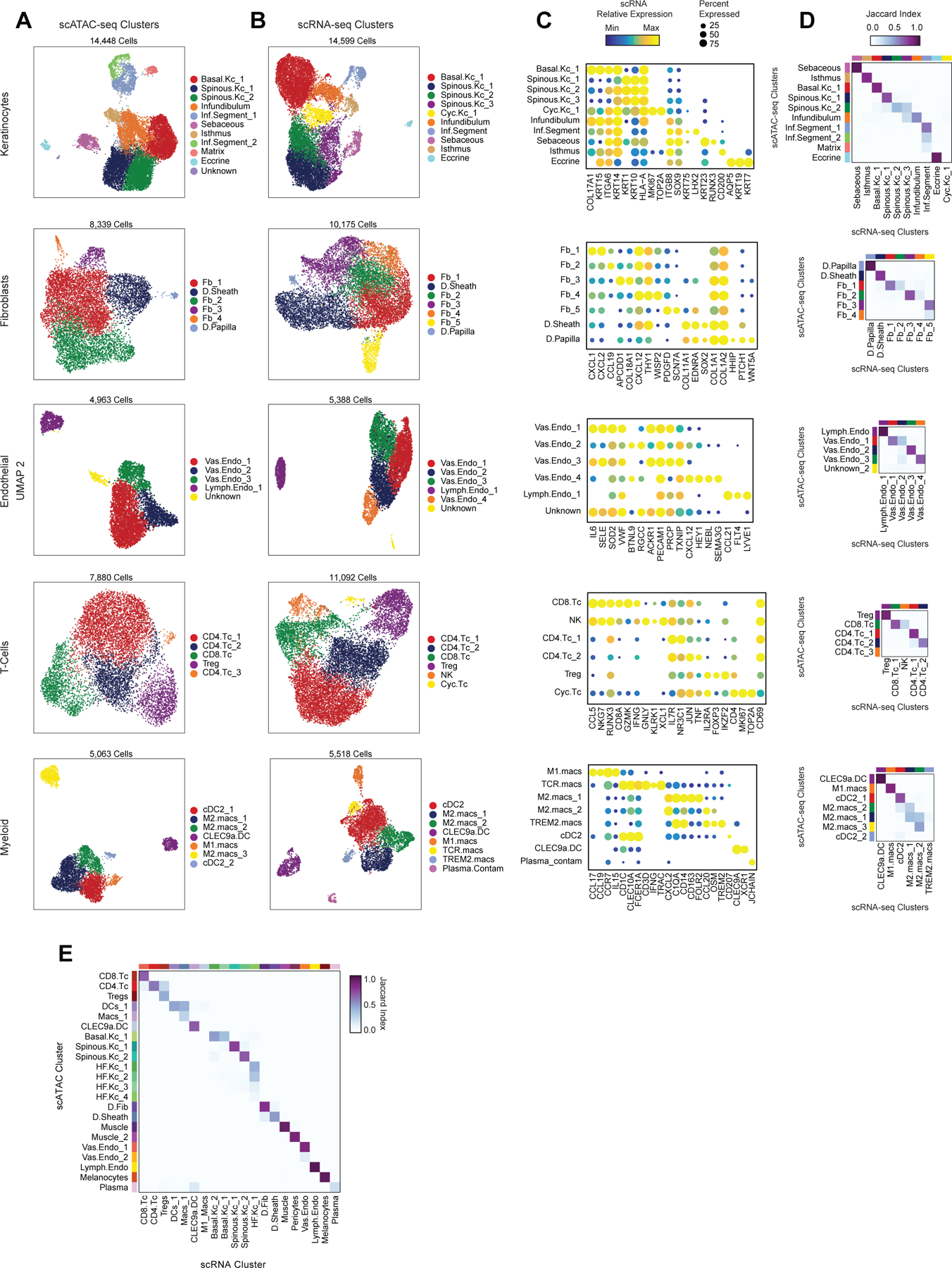
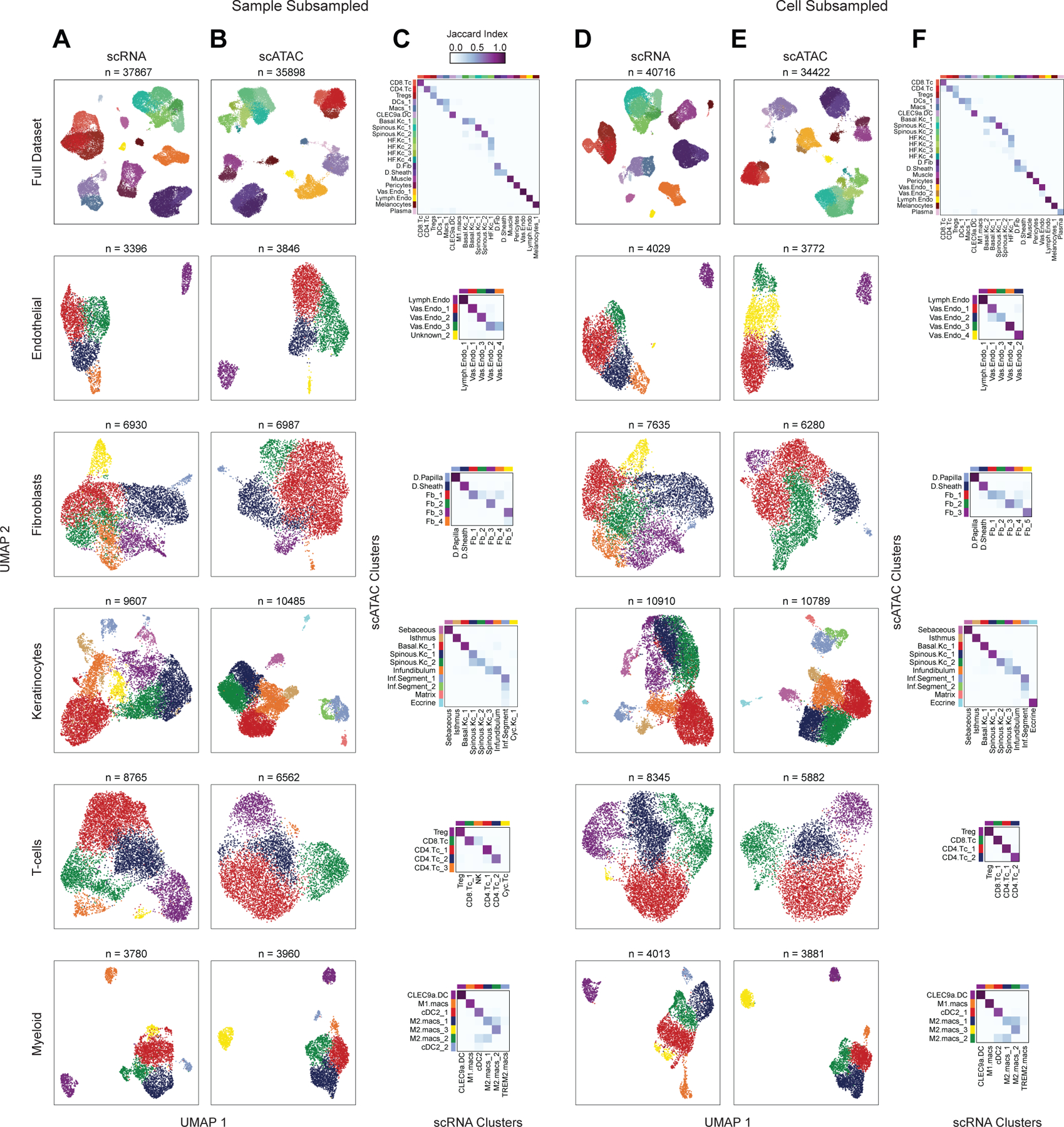
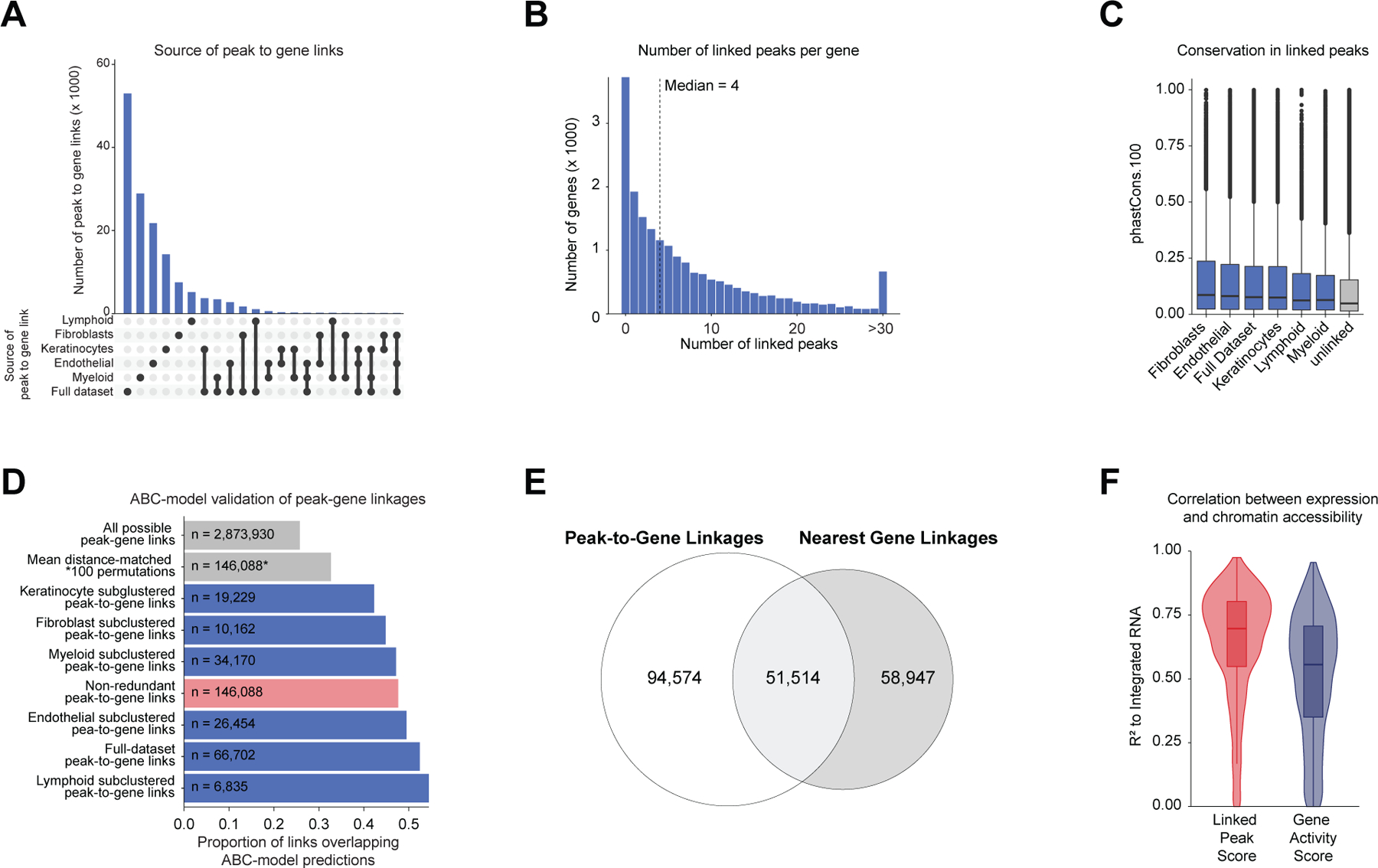
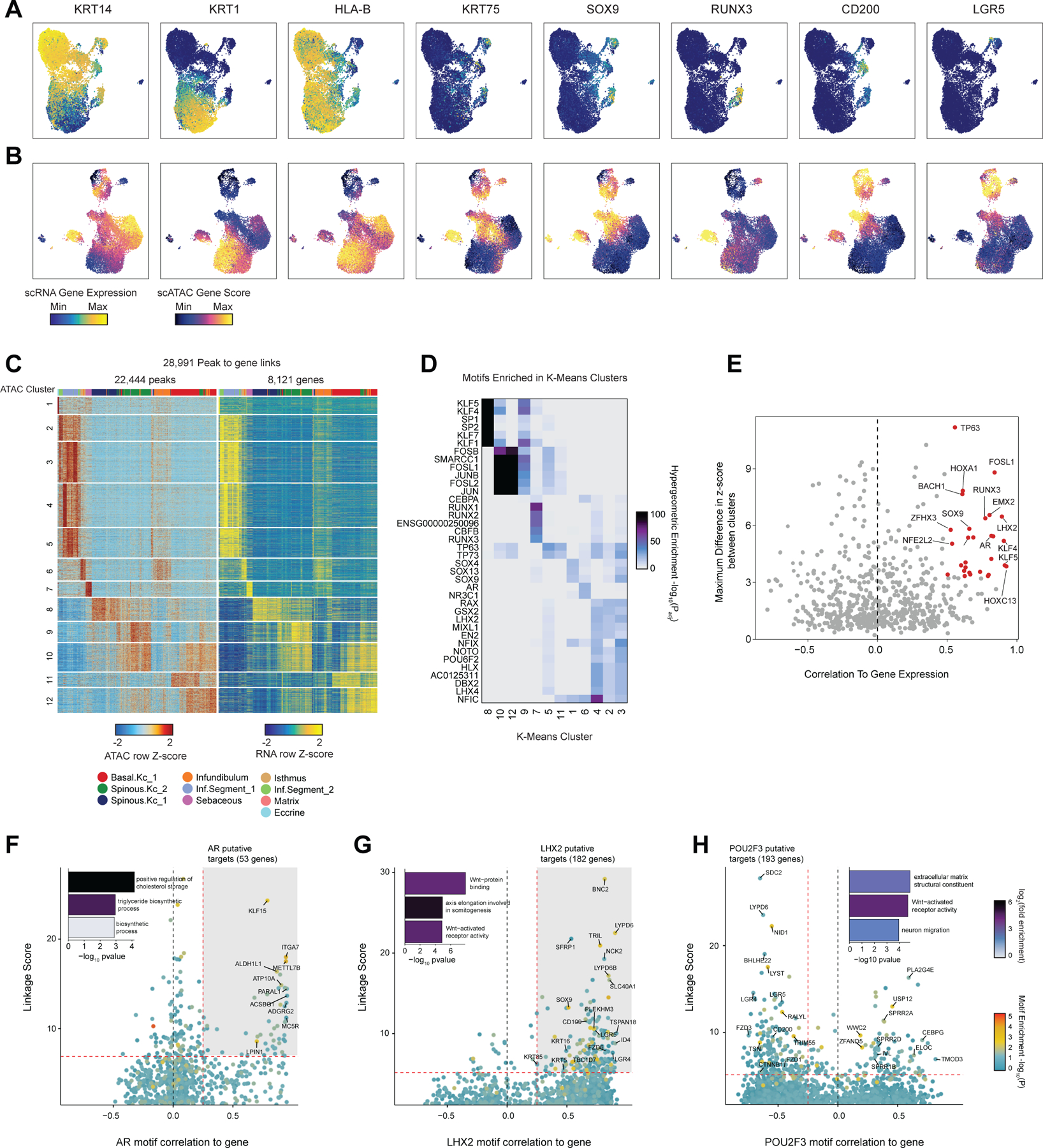
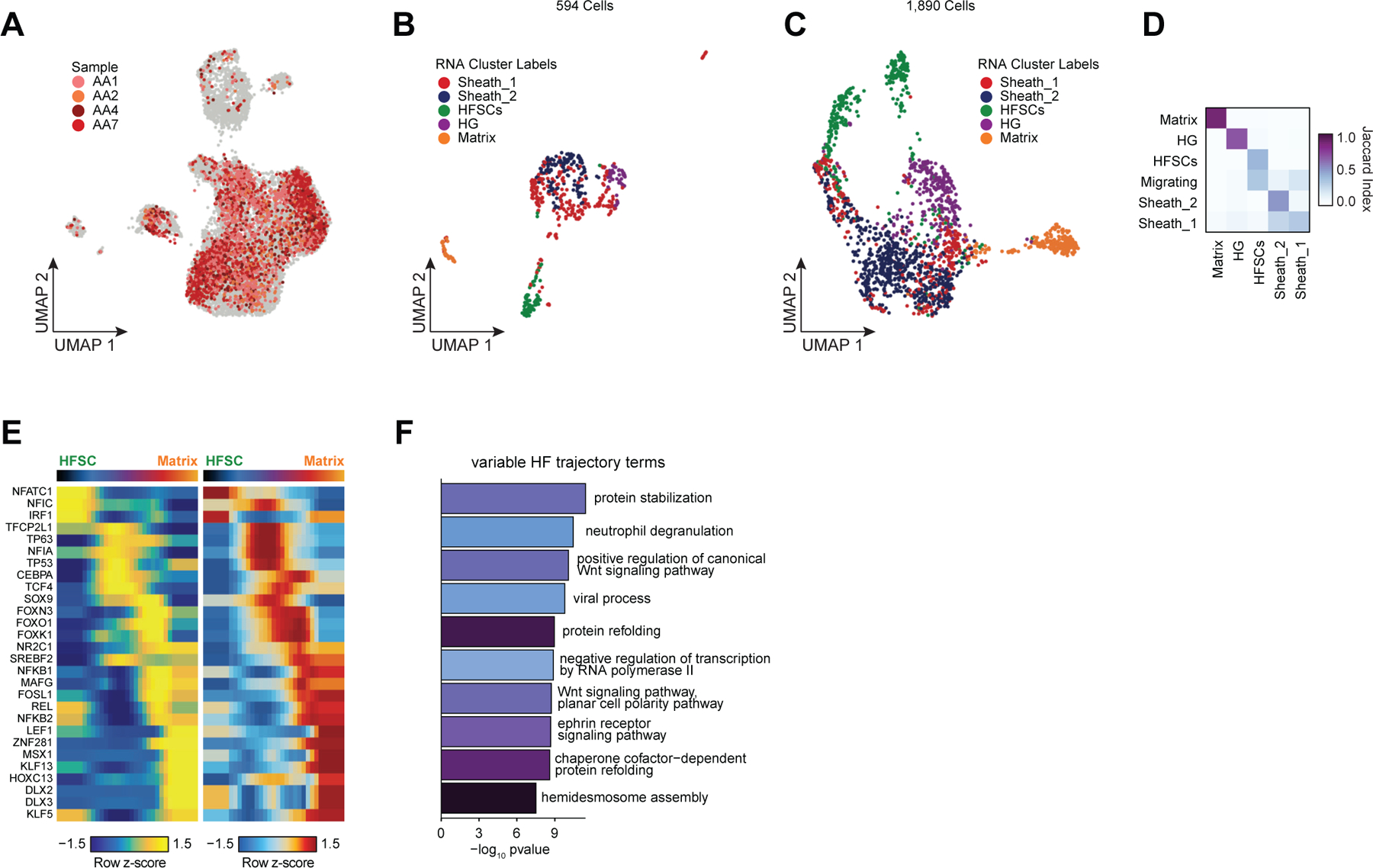
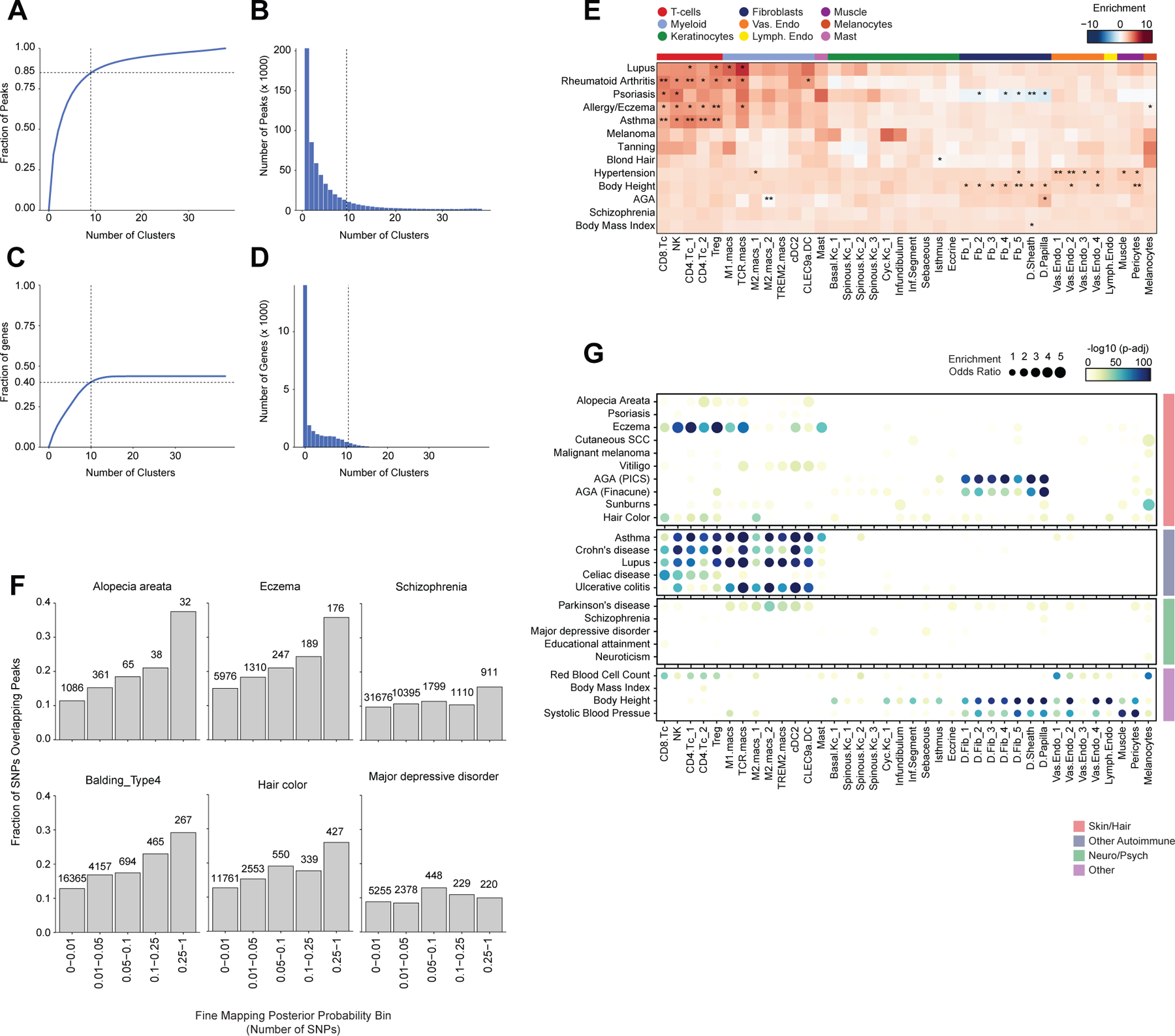
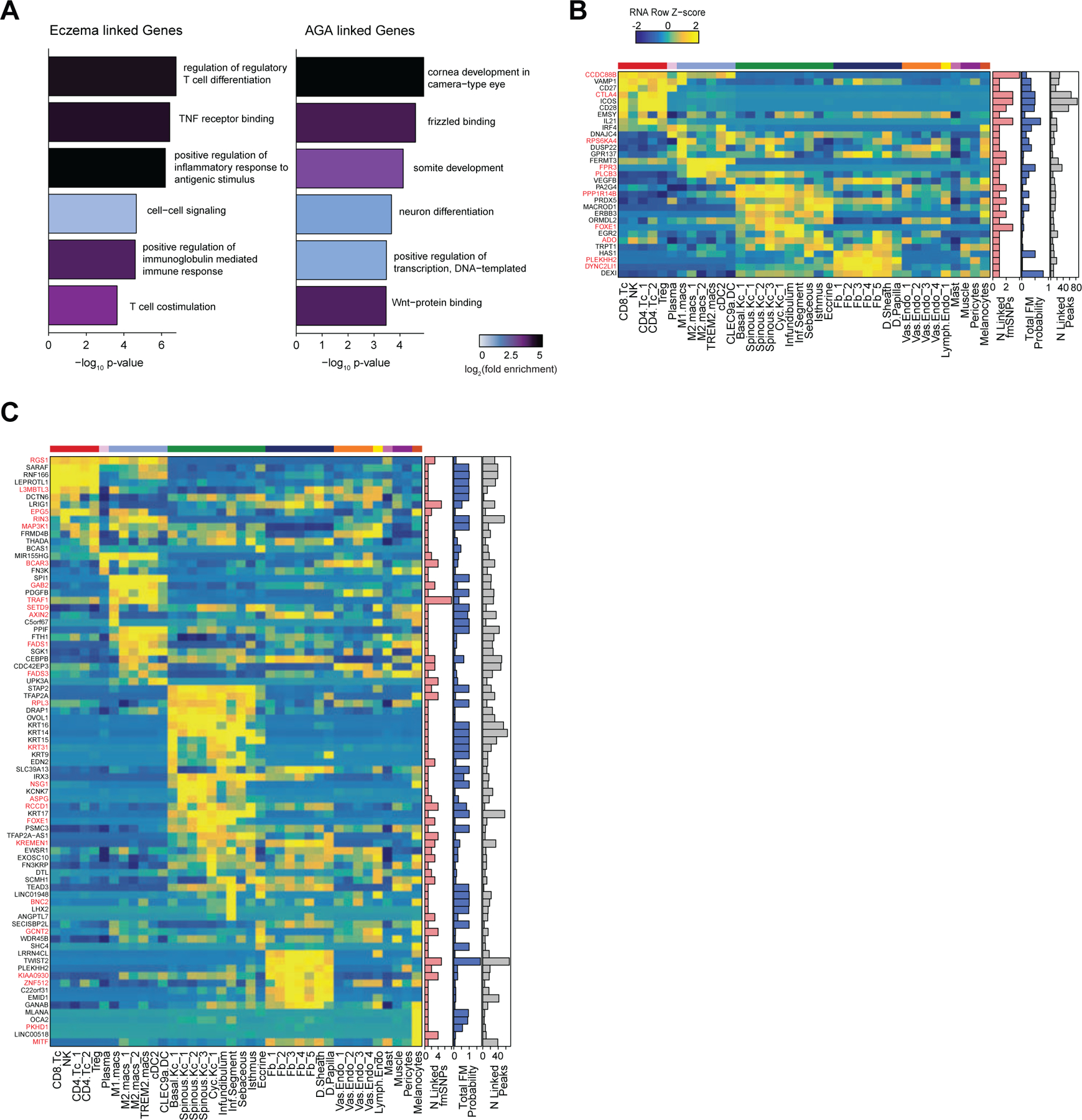
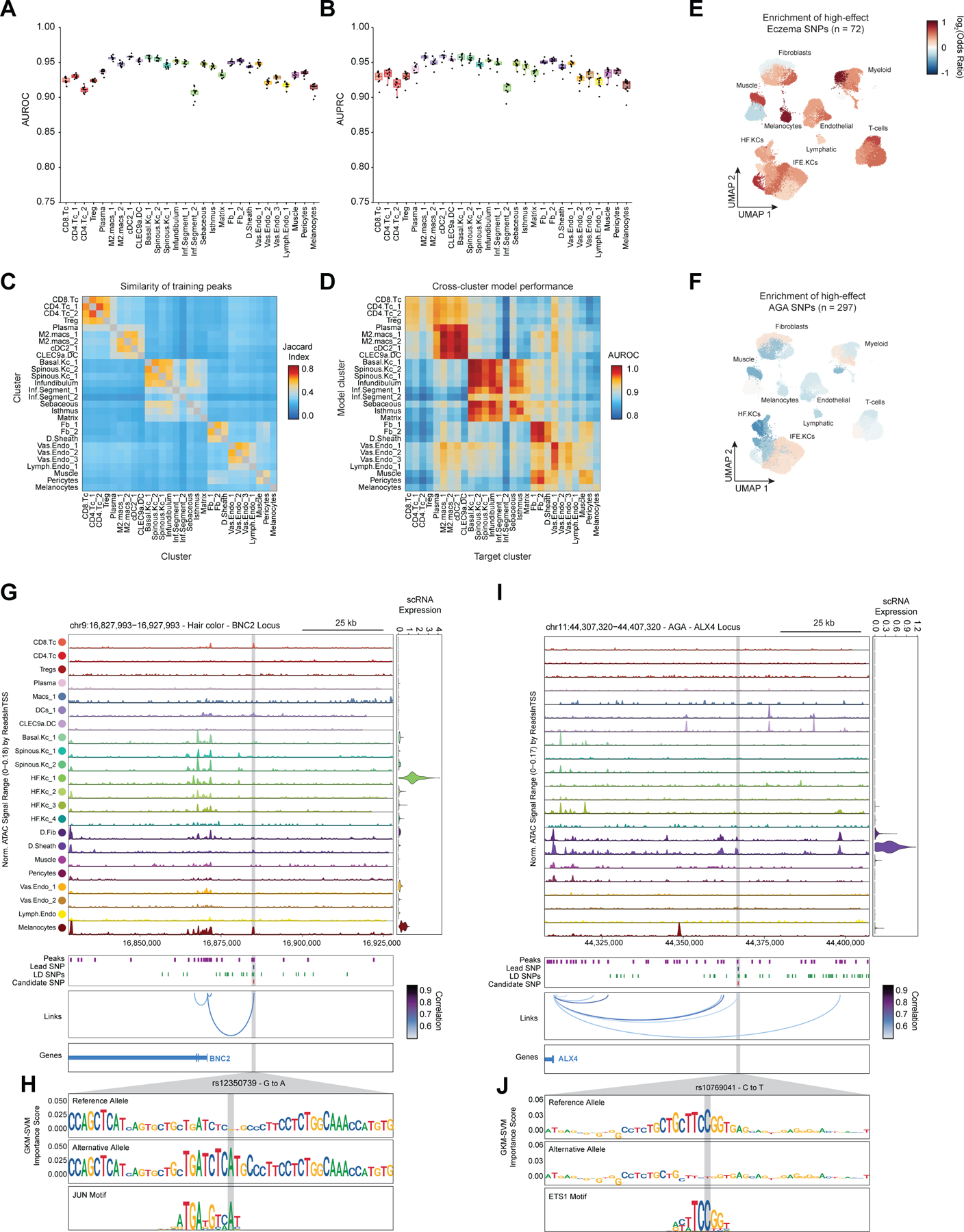
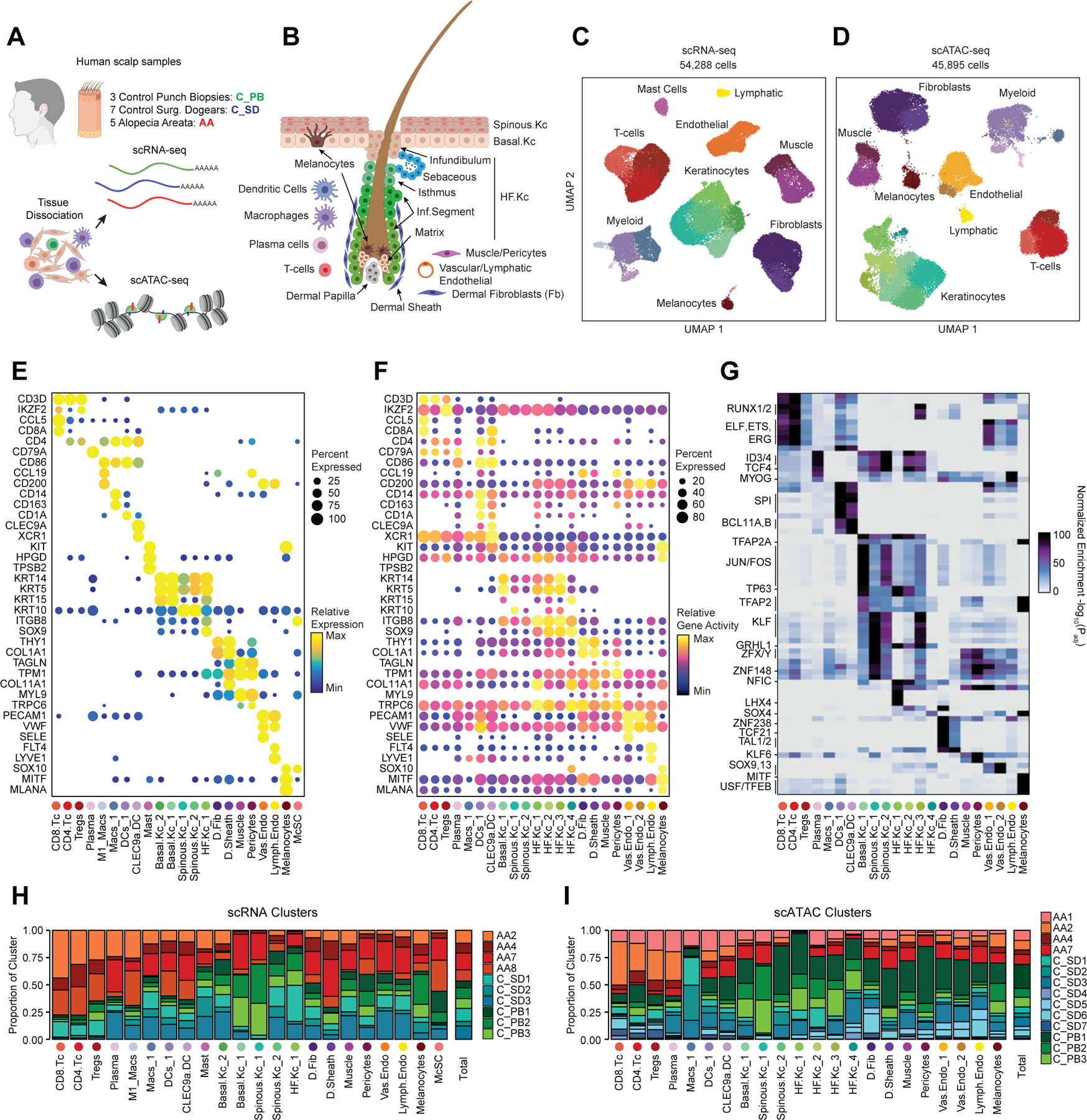
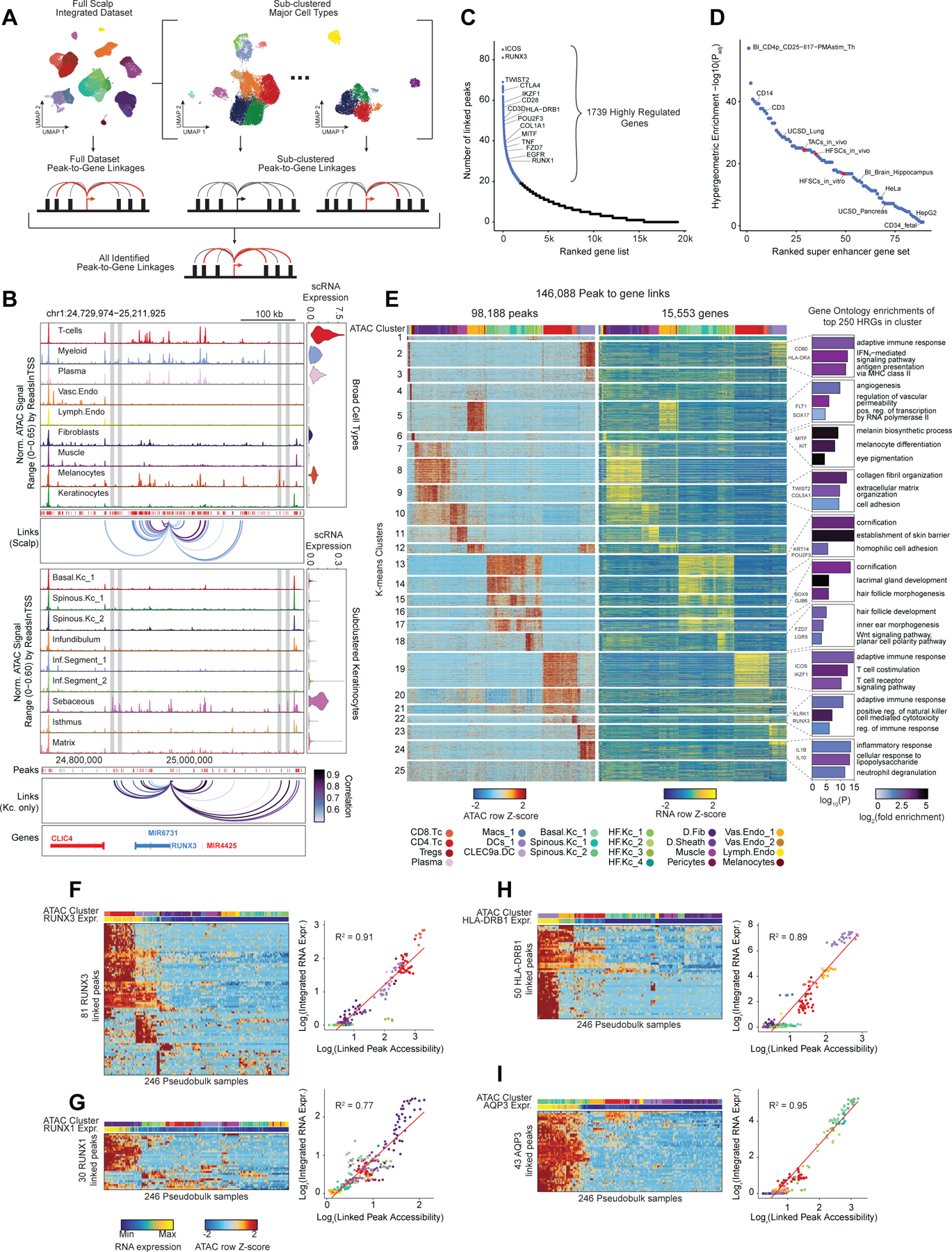
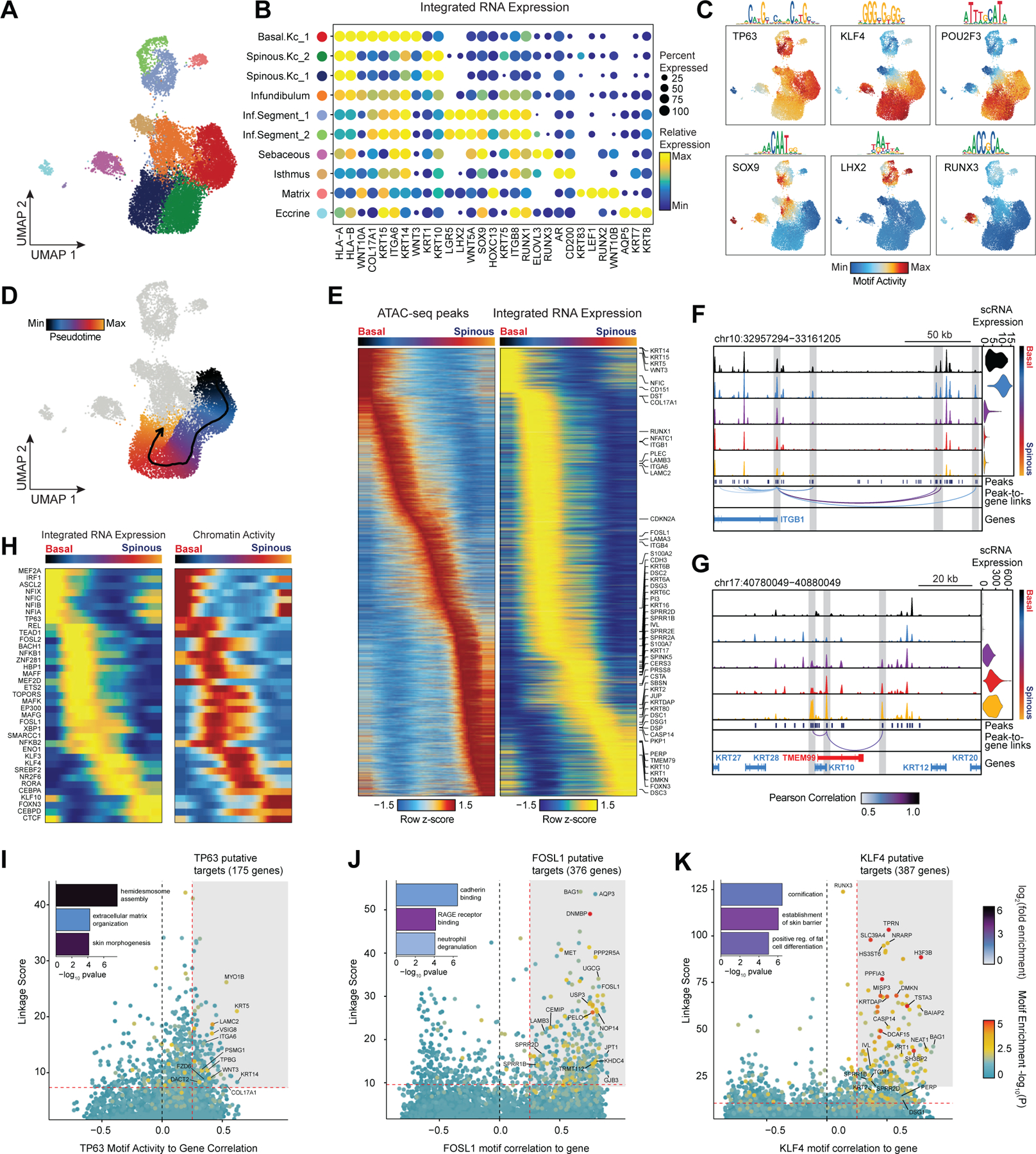
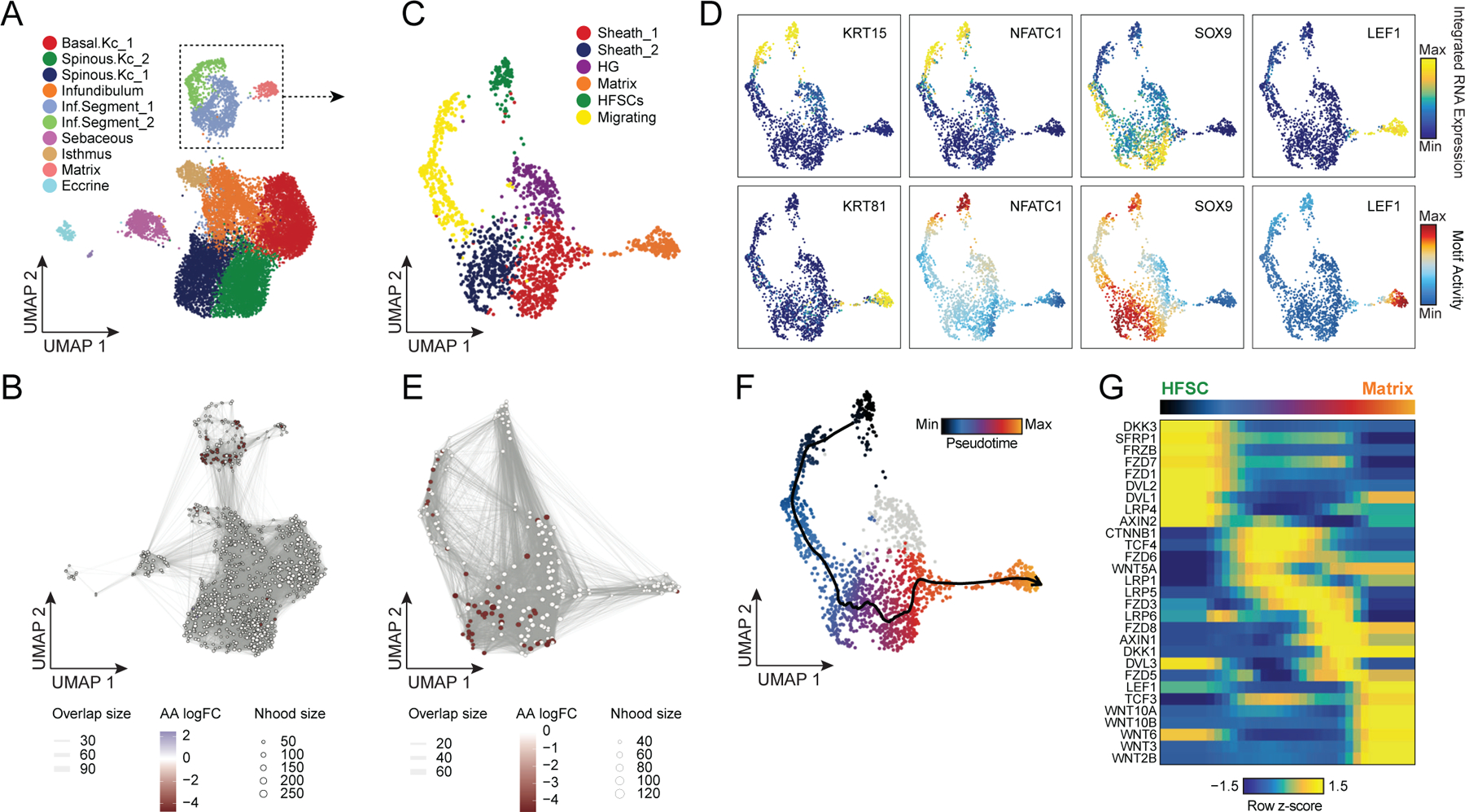
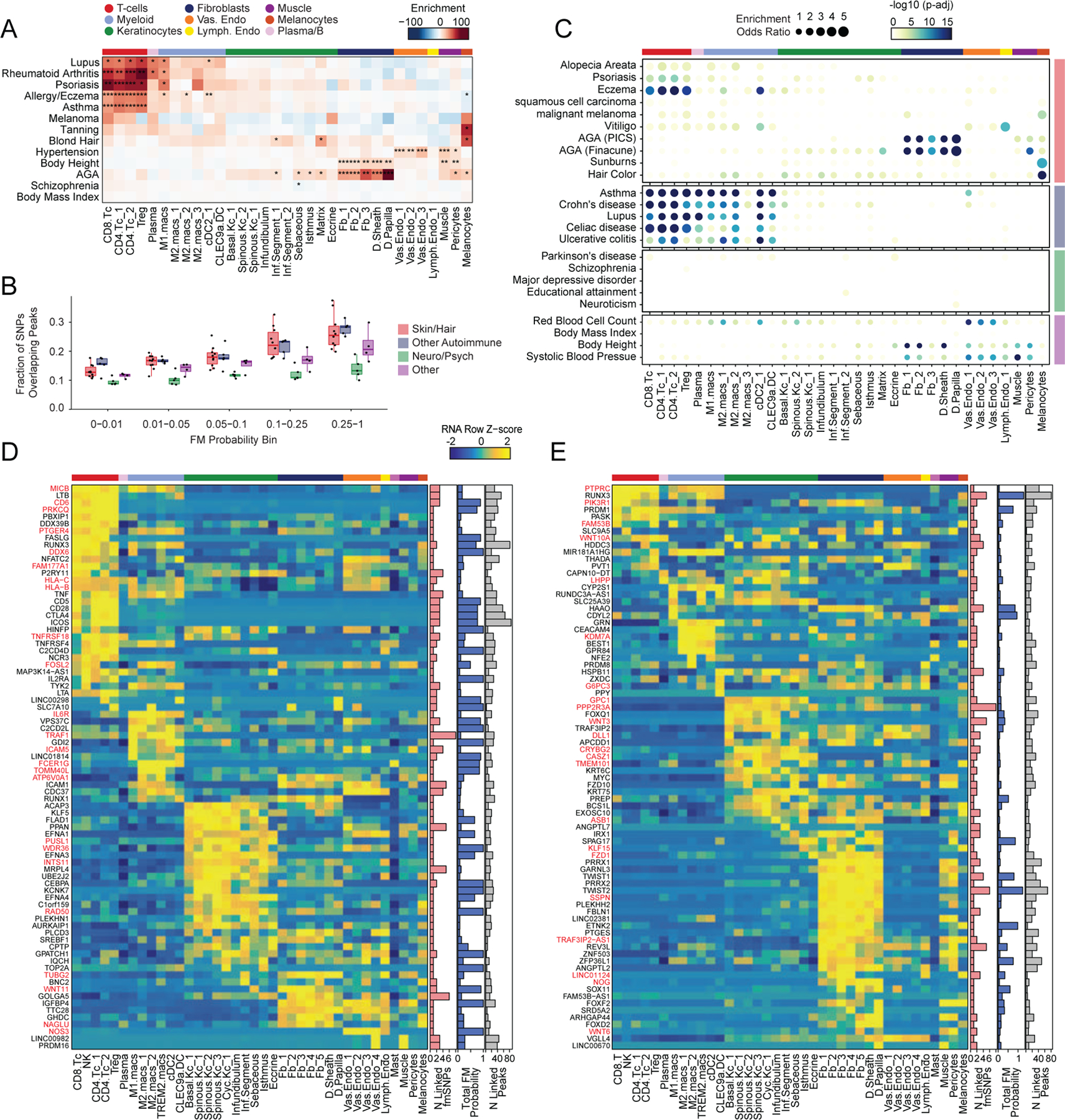
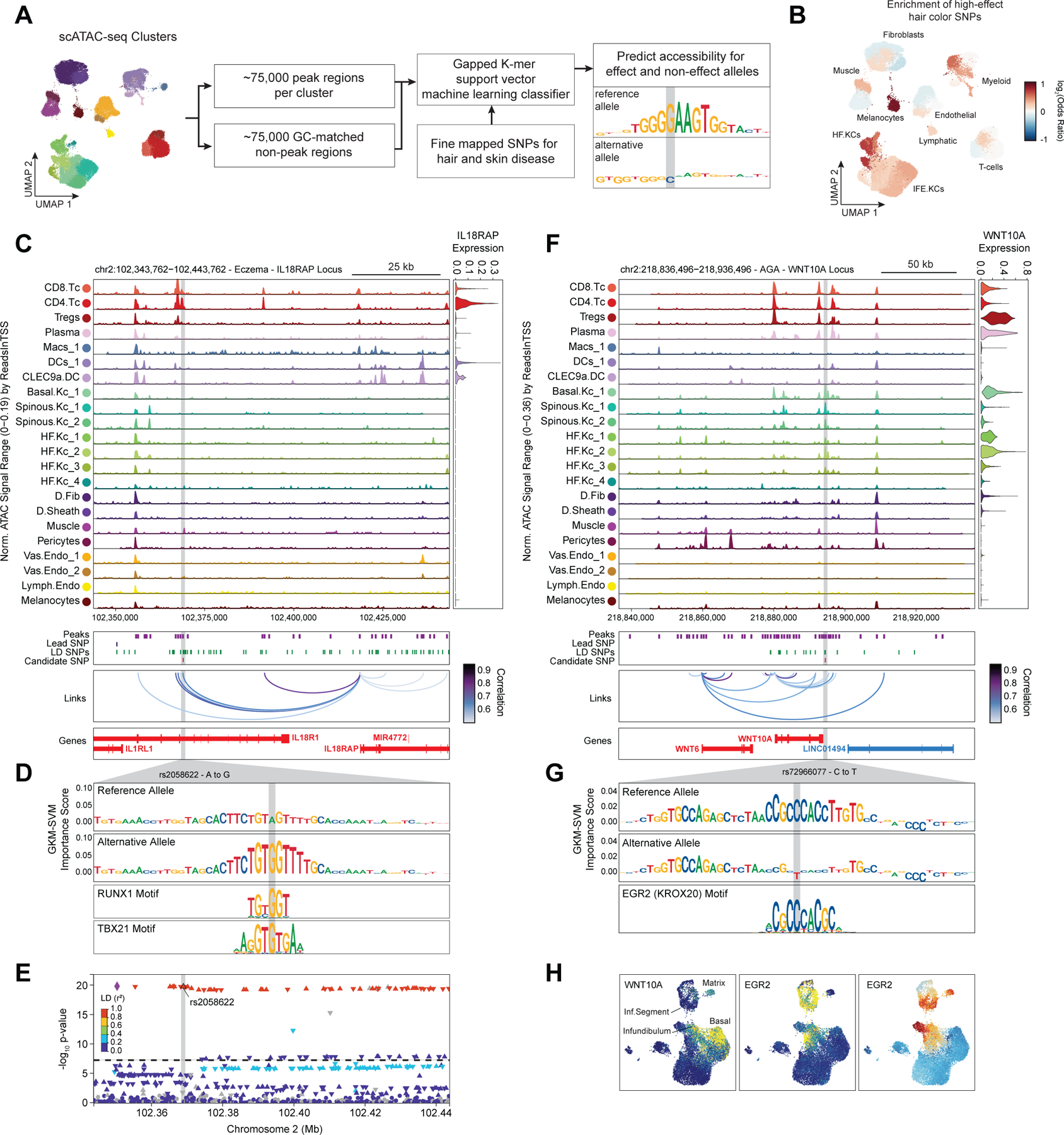
Genome-wide association studies have identified many loci associated with hair and skin disease, but identification of causal variants requires deciphering of gene-regulatory networks in relevant cell types. We generated matched single-cell chromatin profiles and transcriptomes from scalp tissue from healthy controls and patients with alopecia areata, identifying diverse cell types of the hair follicle niche. By interrogating these datasets at multiple levels of cellular resolution, we infer 50-100% more enhancer-gene links than previous approaches and show that aggregate enhancer accessibility for highly regulated genes predicts expression. We use these gene-regulatory maps to prioritize cell types, genes and causal variants implicated in the pathobiology of androgenetic alopecia (AGA), eczema and other complex traits. AGA genome-wide association studies signals are enriched in dermal papilla regulatory regions, supporting the role of these cells as drivers of AGA pathogenesis. Finally, we train machine learning models to nominate single-nucleotide polymorphisms that affect gene expression through disruption of transcription factor binding, predicting candidate functional single-nucleotide polymorphism for AGA and eczema.
© 2023. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Declaration of Interests
W.J.G. is a consultant for 10x Genomics, Guardant Health, Quantapore, Erudio Bio., Lamar Health, Co-founder of Protillion Biosciences, and is named on patents describing ATAC-seq.
Figures

Comment in
-
Identifying critical cell types and gene regulatory pathways for hair and skin disease.Nat Genet. 2023 Aug;55(8):1263-1264. doi: 10.1038/s41588-023-01447-2. Nat Genet. 2023. PMID: 37500732 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
- RM1 HG007735/HG/NHGRI NIH HHS/United States
- T32 GM007365/GM/NIGMS NIH HHS/United States
- T32 GM145402/GM/NIGMS NIH HHS/United States
- U19 AI057266/AI/NIAID NIH HHS/United States
- R01 HG009909/HG/NHGRI NIH HHS/United States
- U01 HG011762/HG/NHGRI NIH HHS/United States
- P50 HG007735/HG/NHGRI NIH HHS/United States
- UM1 HG011972/HG/NHGRI NIH HHS/United States
- R01 NS128028/NS/NINDS NIH HHS/United States
- U54 HG012723/HG/NHGRI NIH HHS/United States
- UM1 HG009442/HG/NHGRI NIH HHS/United States
- R37 AR054780/AR/NIAMS NIH HHS/United States
- P01 AI153559/AI/NIAID NIH HHS/United States
- UM1 HG009436/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
