

Single-molecule genome-wide mutation profiles of cell-free DNA for non-invasive detection of cancer
- PMID: 37500728
- PMCID: PMC10412448
- DOI: 10.1038/s41588-023-01446-3
Single-molecule genome-wide mutation profiles of cell-free DNA for non-invasive detection of cancer
Abstract
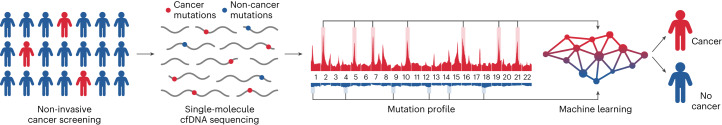
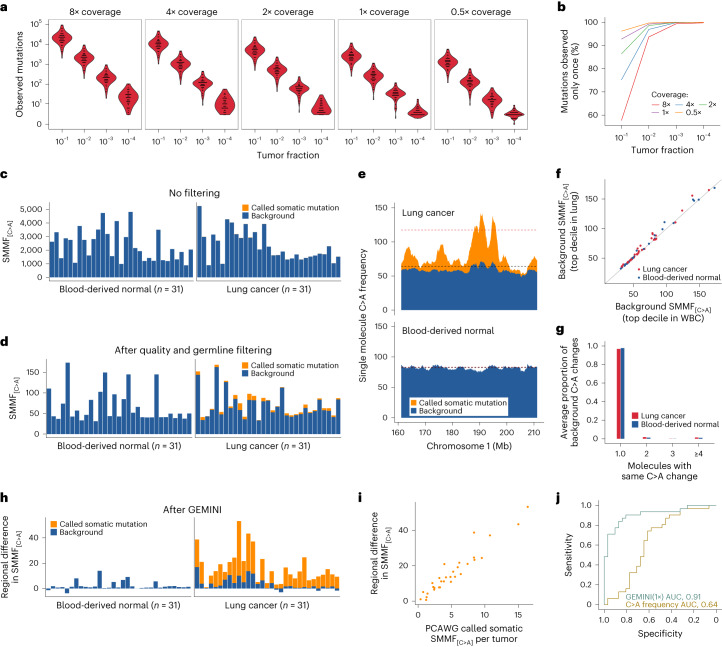
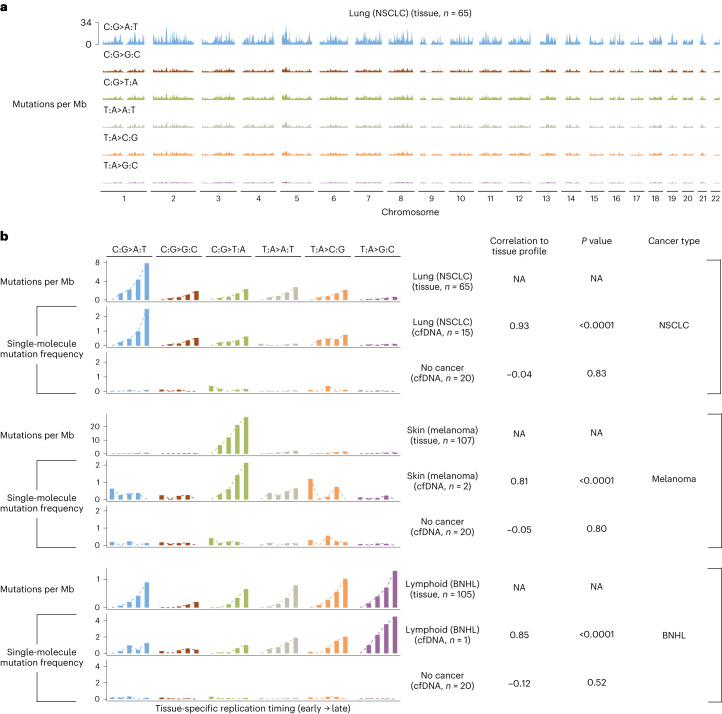
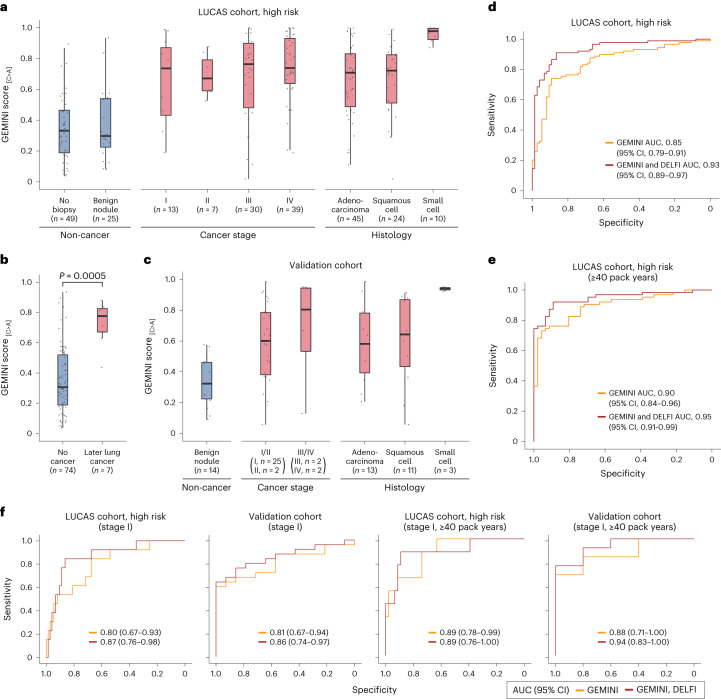
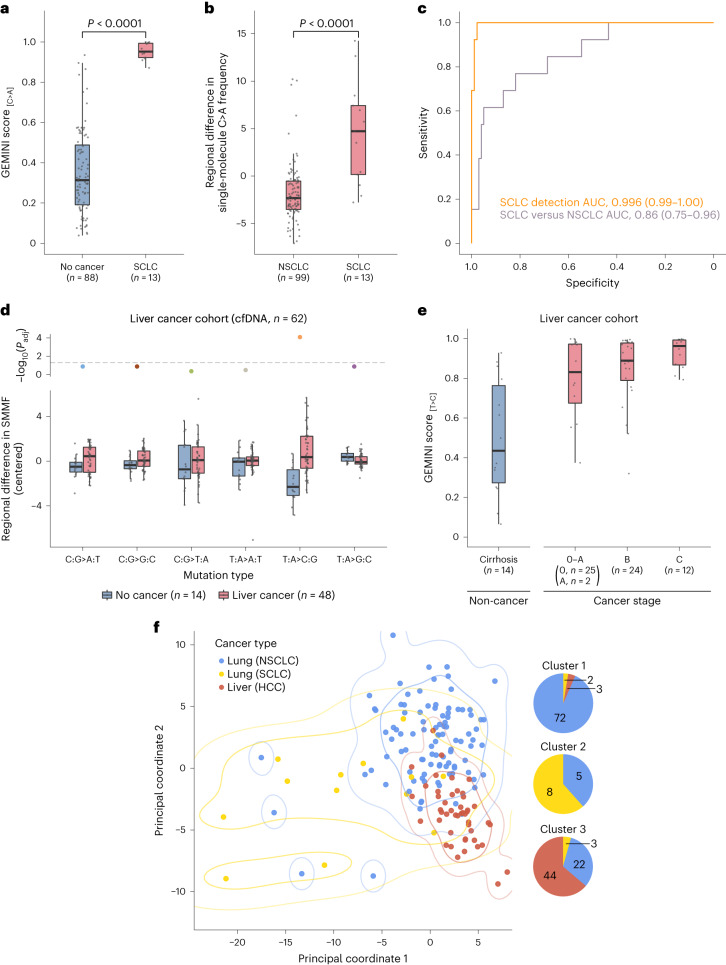
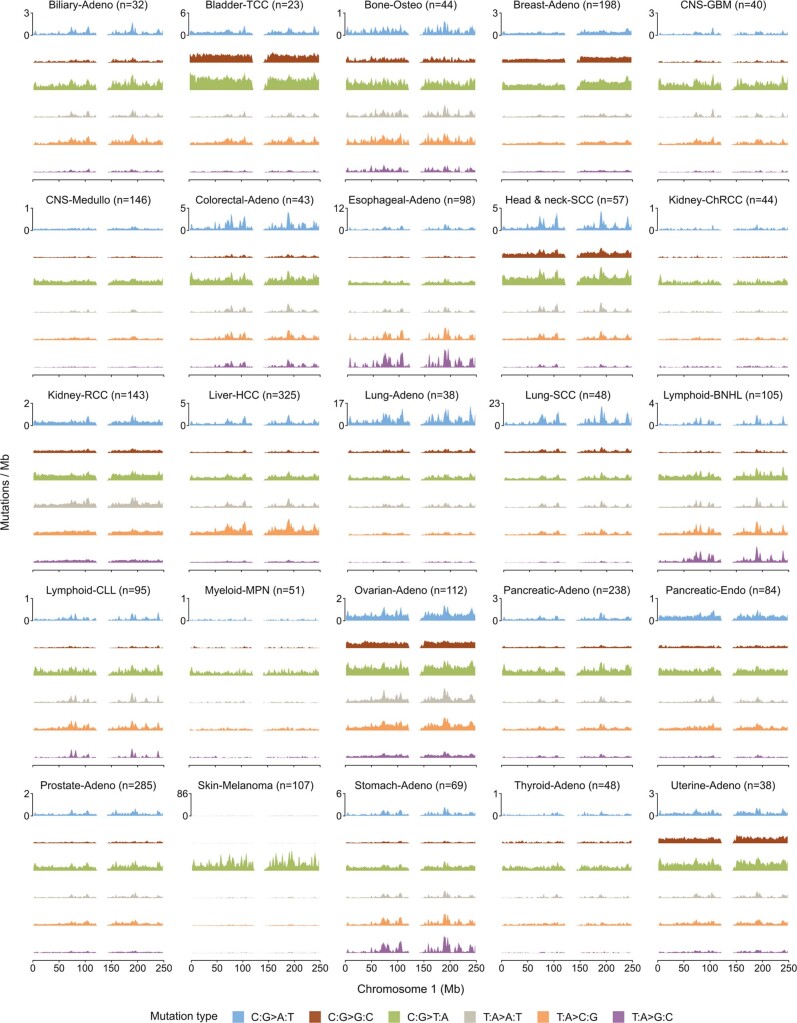
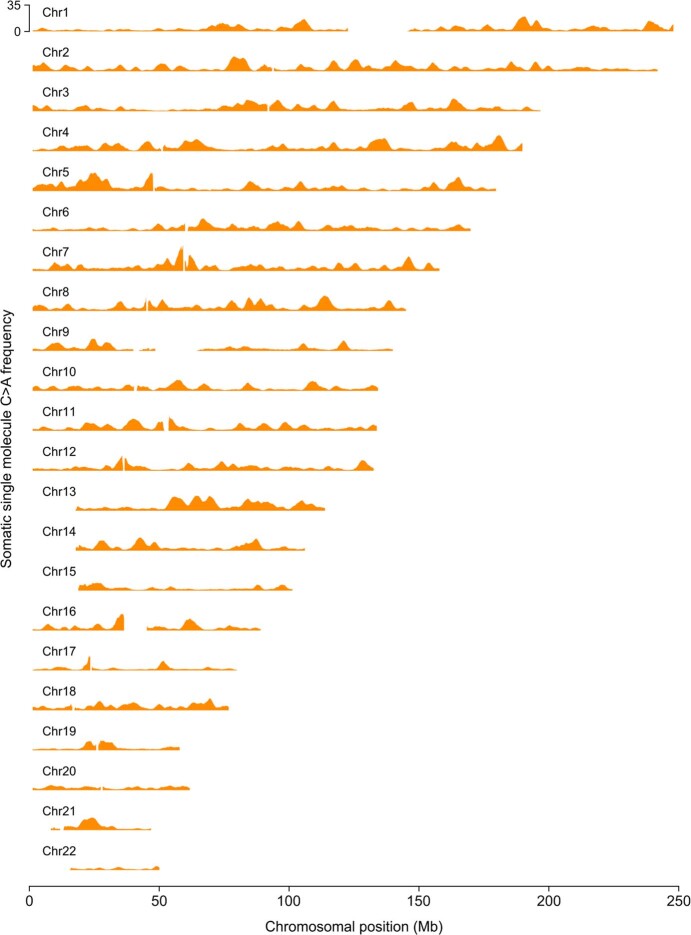
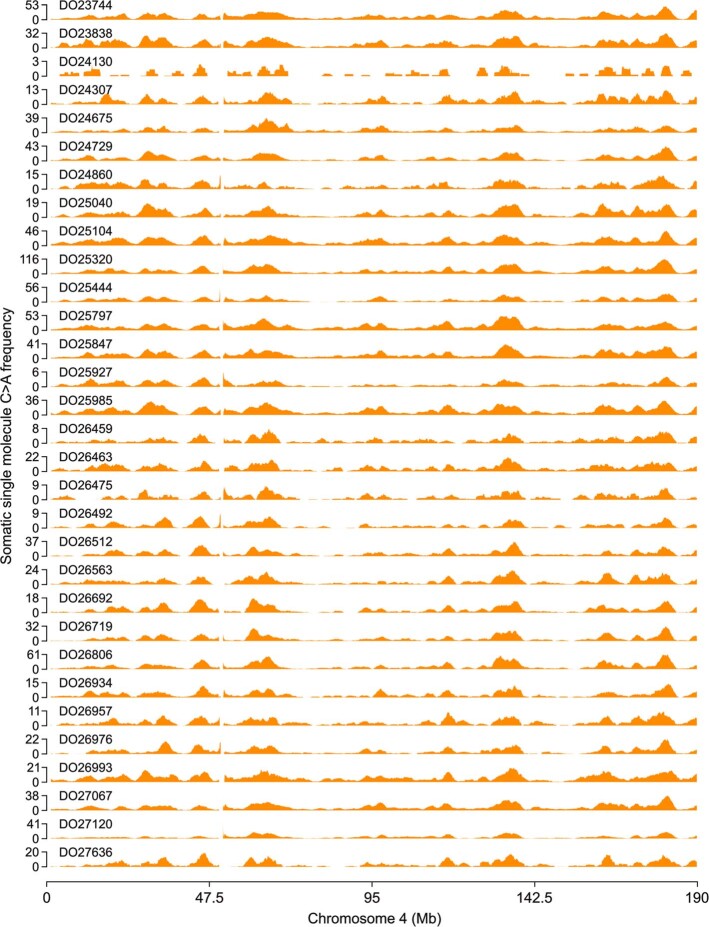
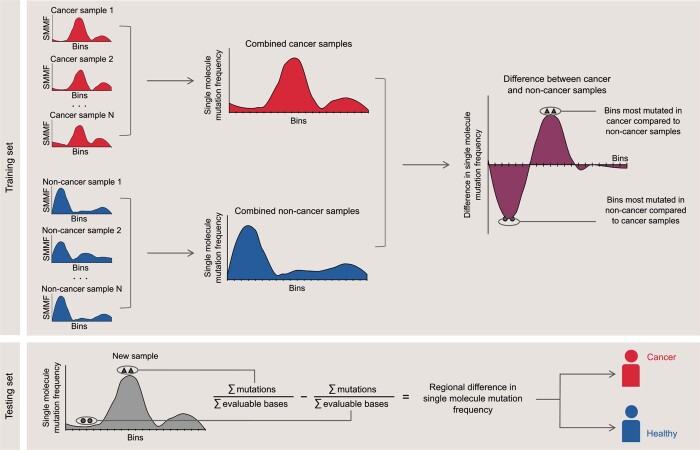
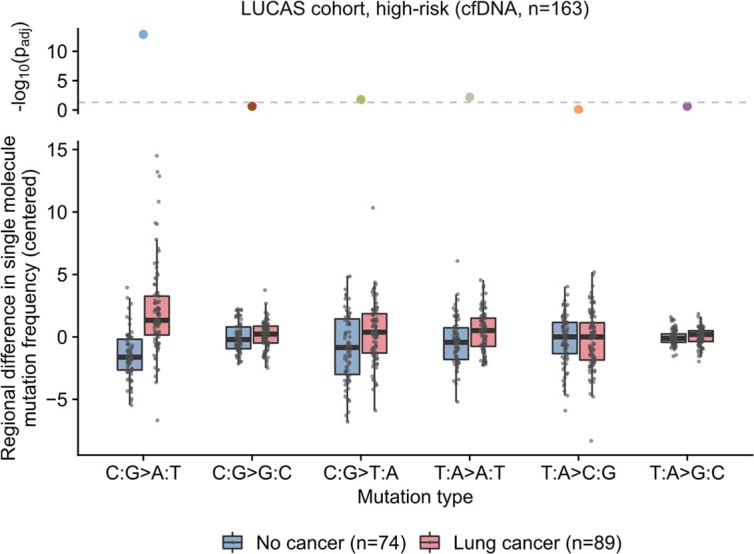
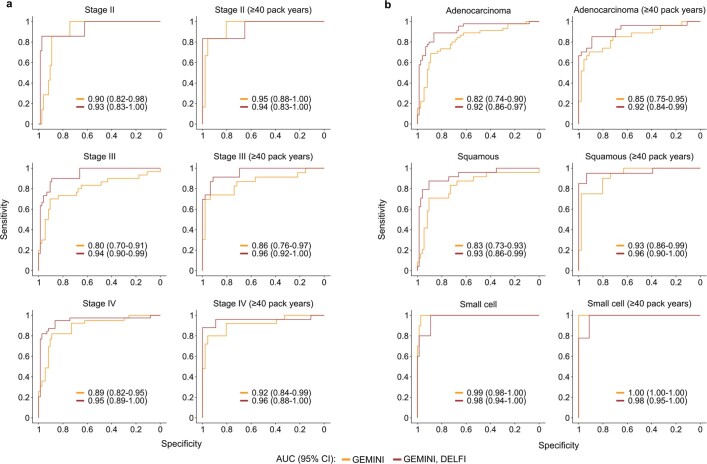
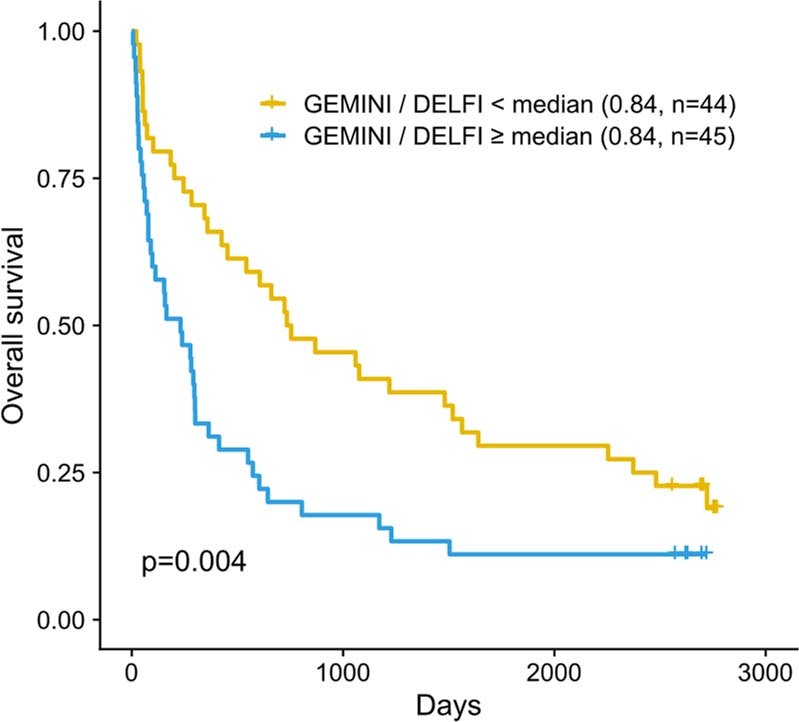
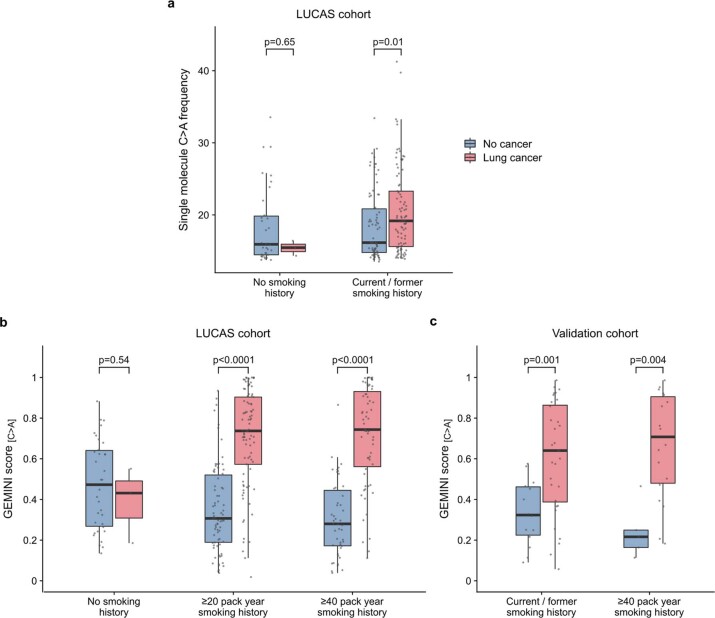
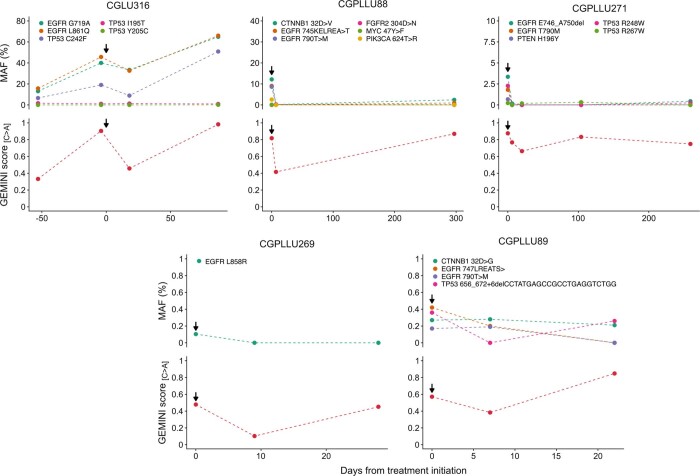
Somatic mutations are a hallmark of tumorigenesis and may be useful for non-invasive diagnosis of cancer. We analyzed whole-genome sequencing data from 2,511 individuals in the Pan-Cancer Analysis of Whole Genomes (PCAWG) study as well as 489 individuals from four prospective cohorts and found distinct regional mutation type-specific frequencies in tissue and cell-free DNA from patients with cancer that were associated with replication timing and other chromatin features. A machine-learning model using genome-wide mutational profiles combined with other features and followed by CT imaging detected >90% of patients with lung cancer, including those with stage I and II disease. The fixed model was validated in an independent cohort, detected patients with cancer earlier than standard approaches and could be used to monitor response to therapy. This approach lays the groundwork for non-invasive cancer detection using genome-wide mutation features that may facilitate cancer screening and monitoring.
© 2023. The Author(s).
Conflict of interest statement
D.C.B., D.M., S.C., V. Adleff, J.P., V. Anagnostou, R.B.S. and V.E.V. are inventors on patent applications submitted by Johns Hopkins University related to cfDNA for cancer detection. S.C., J.P., V. Adleff. and R.B.S. are founders of Delfi Diagnostics, and V. Adleff. and R.B.S. are consultants for this organization. J.R.W. is the founder and owner of Resphera Biosciences. V.E.V. is a founder of Delfi Diagnostics, serves on the Board of Directors and as an officer for this organization and owns Delfi Diagnostics stock, which is subject to certain restrictions under university policy. Additionally, Johns Hopkins University owns equity in Delfi Diagnostics. V.E.V. divested his equity in Personal Genome Diagnostics (PGDx) to LabCorp in February 2022. V.E.V. is an inventor on patent applications submitted by Johns Hopkins University related to cancer genomic analyses and cfDNA for cancer detection that have been licensed to one or more entities, including Delfi Diagnostics, LabCorp, Qiagen, Sysmex, Agios, Genzyme, Esoterix, Ventana and ManaT Bio. Under the terms of these license agreements, the University and inventors are entitled to fees and royalty distributions. V.E.V. is an advisor to Viron Therapeutics and Epitope. These arrangements have been reviewed and approved by Johns Hopkins University in accordance with its conflict-of-interest policies. The remaining authors declare no competing interests.
Figures

Comment in
-
Genome-wide mutation profiles from cell-free DNA for early cancer detection.Nat Genet. 2023 Aug;55(8):1261-1262. doi: 10.1038/s41588-023-01448-1. Nat Genet. 2023. PMID: 37524791 No abstract available.
References
-
- Guide to Cancer Early Diagnosis (World Health Organization, 2017).
-
- Moyer VA. U.S. Preventive Services Task Force. Screening for lung cancer: U.S. Preventive Services Task Force recommendation statement. Ann. Intern. Med. 2014;160:330–338. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
