

Multi-omic rejuvenation and life span extension on exposure to youthful circulation
- PMID: 37500973
- PMCID: PMC11095548
- DOI: 10.1038/s43587-023-00451-9
Multi-omic rejuvenation and life span extension on exposure to youthful circulation
Abstract
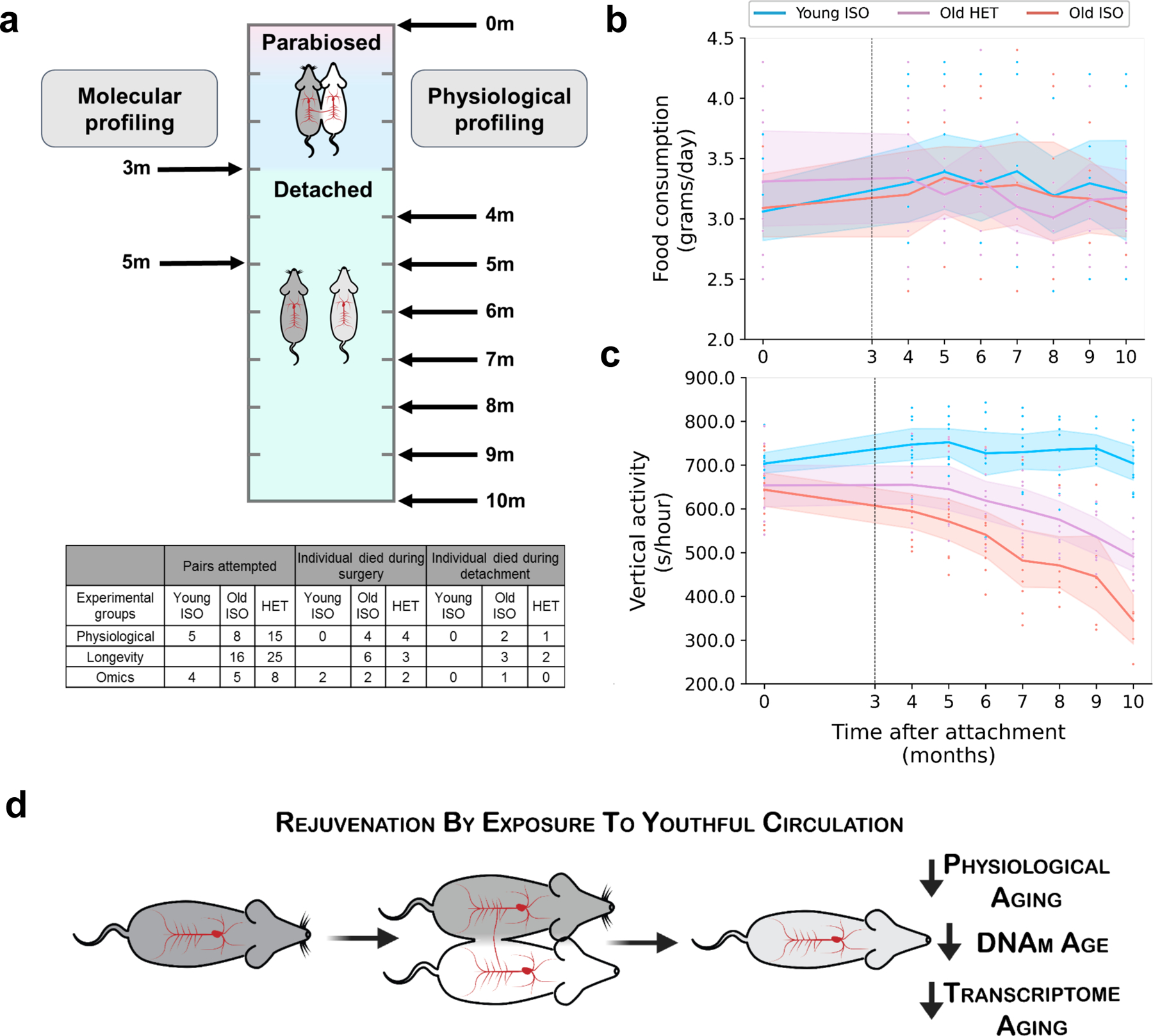
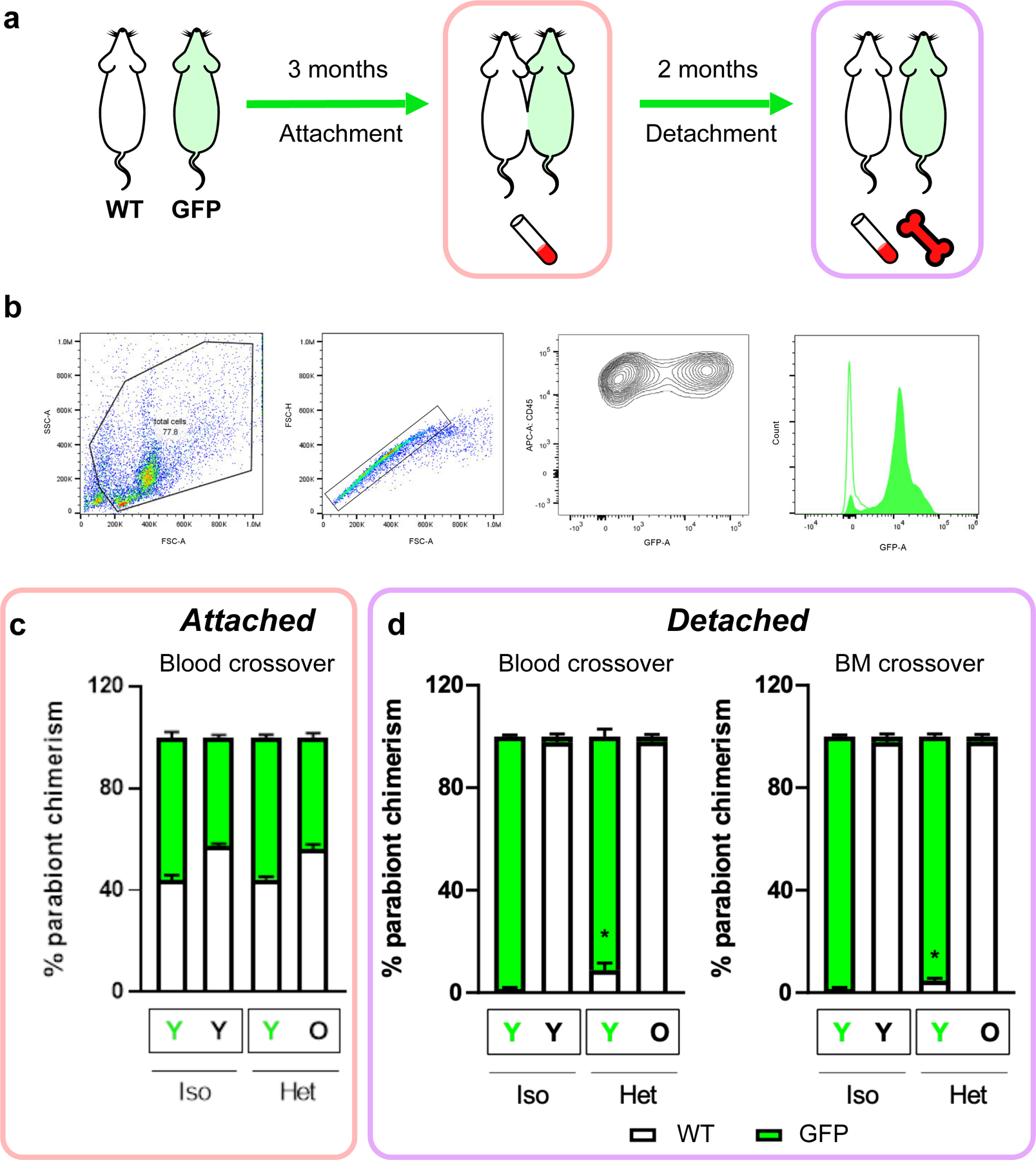
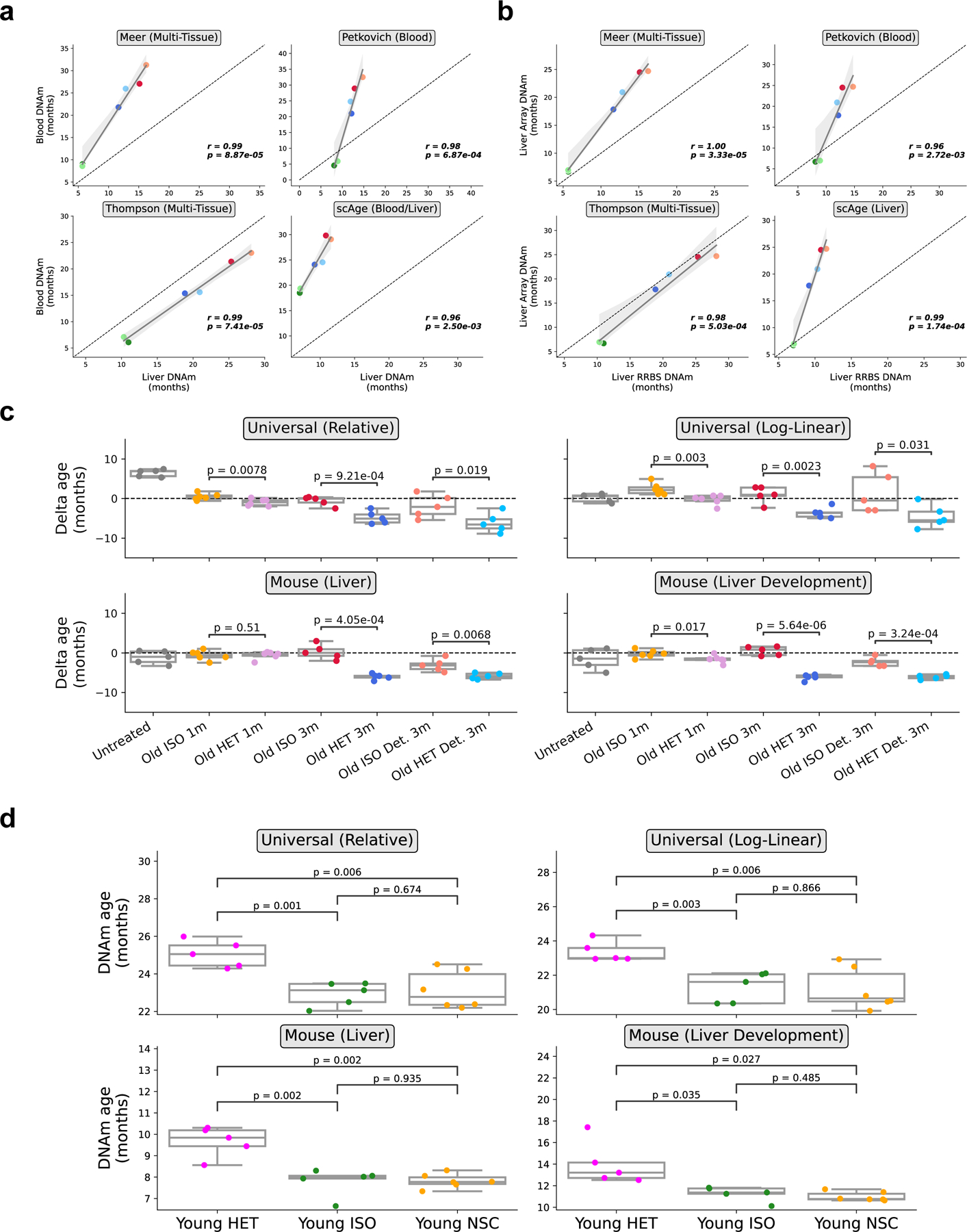
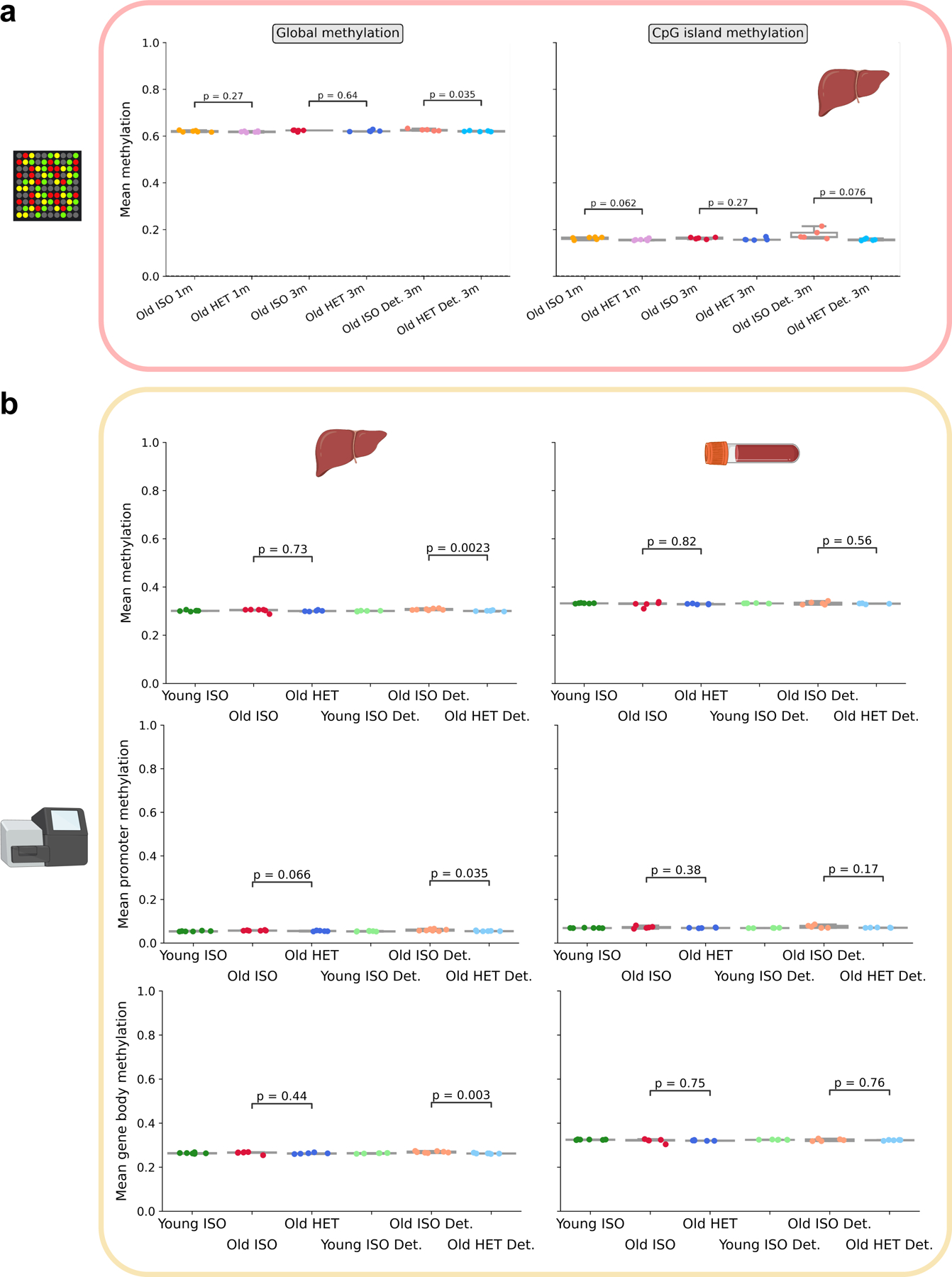
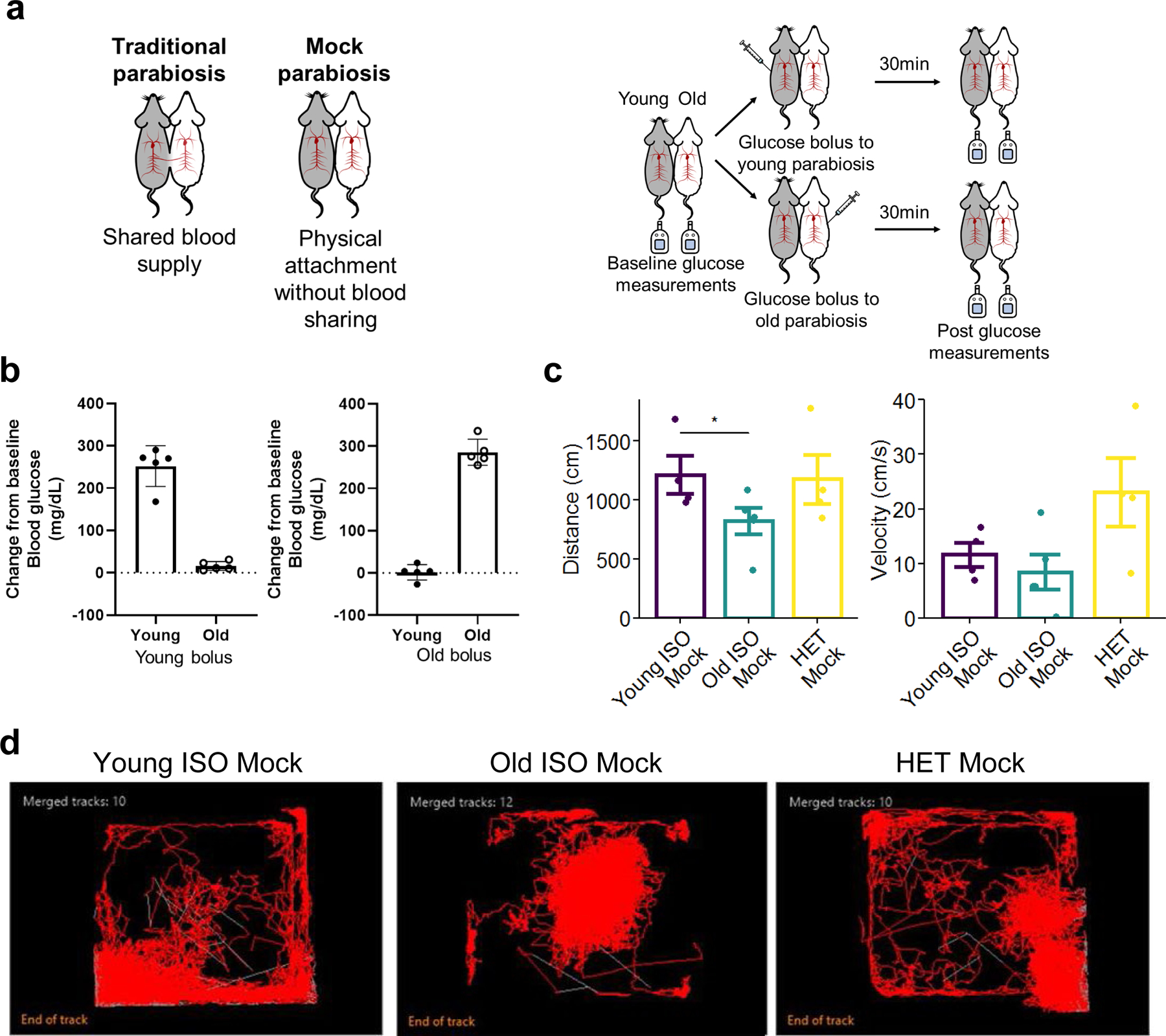
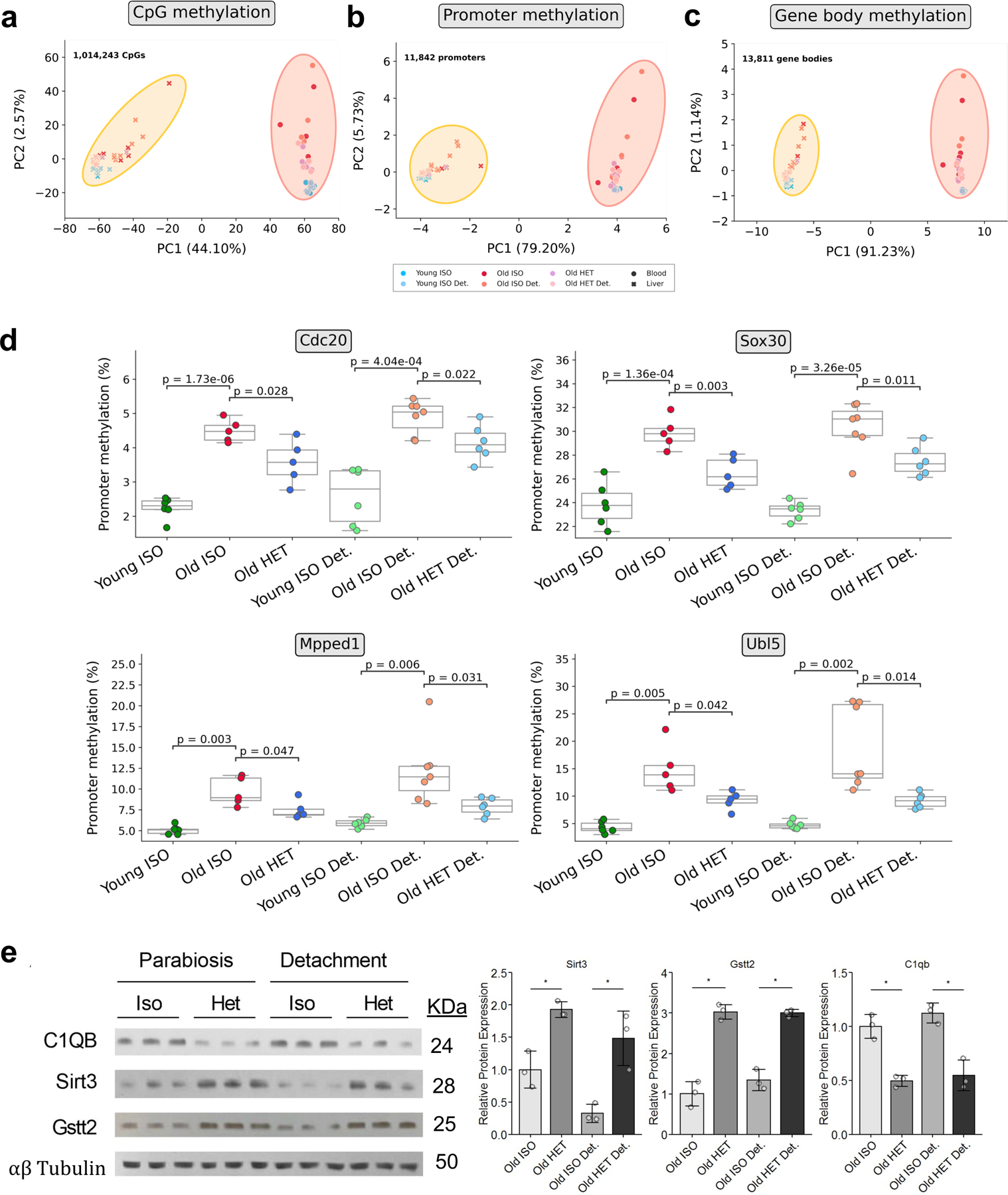
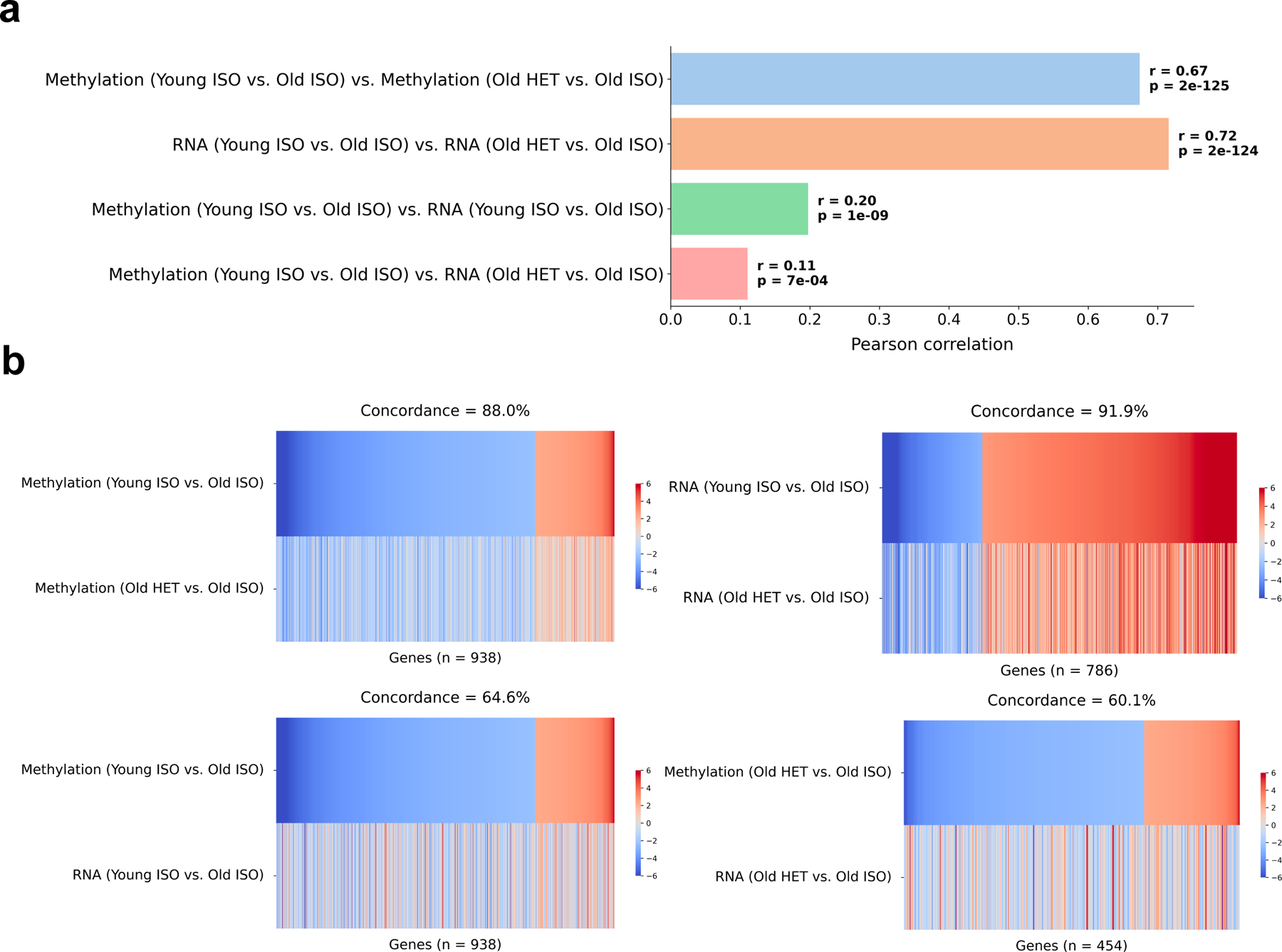
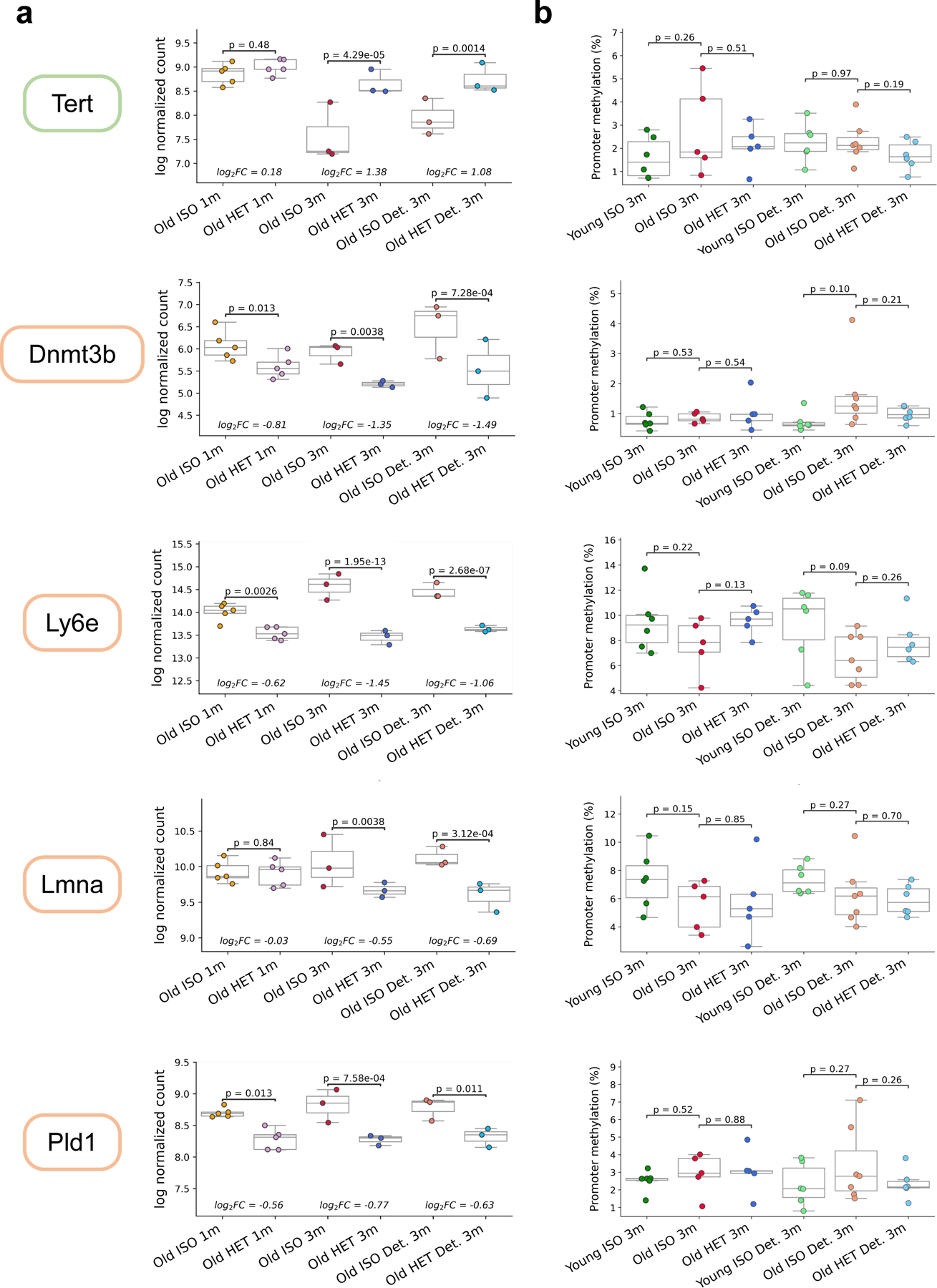
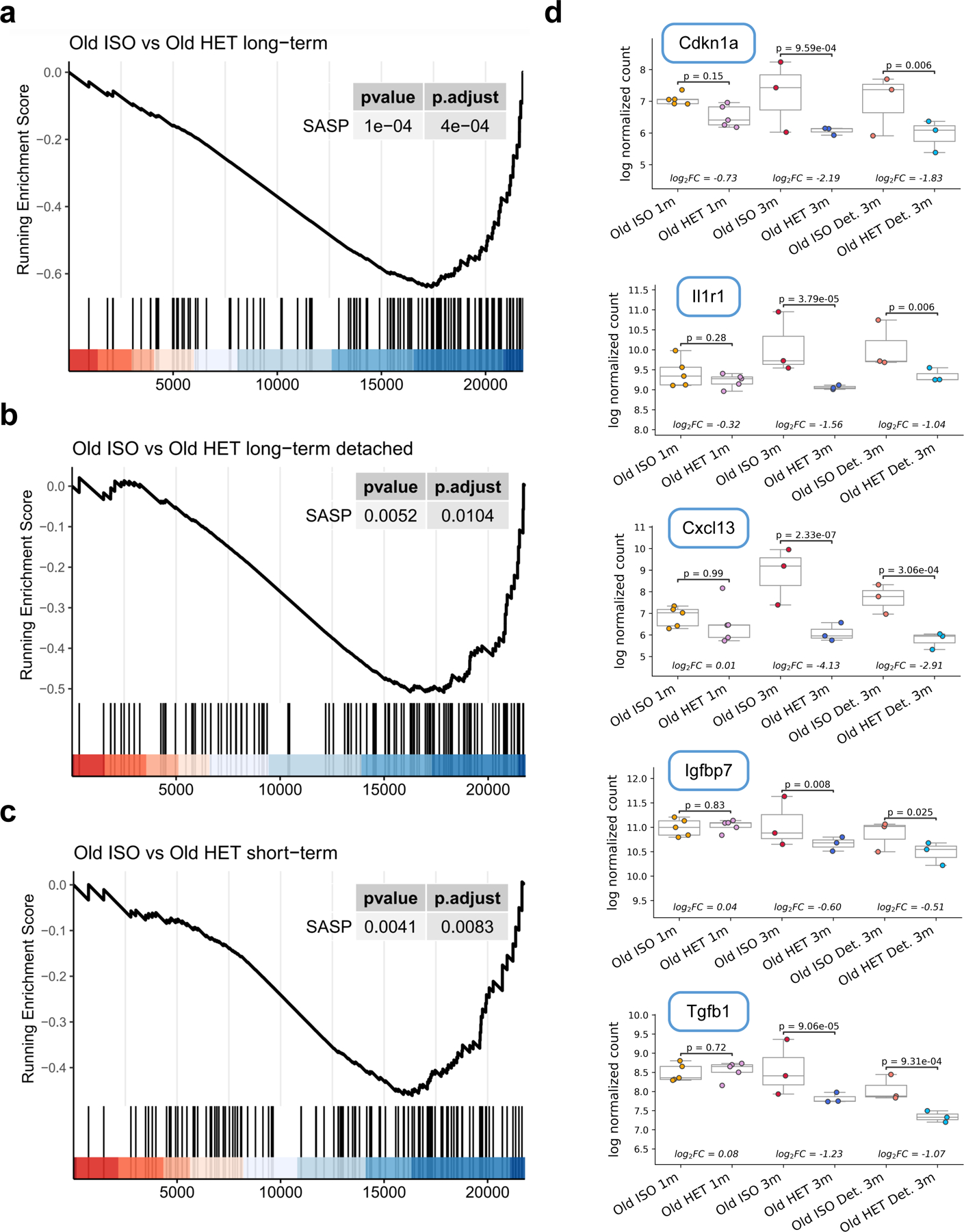
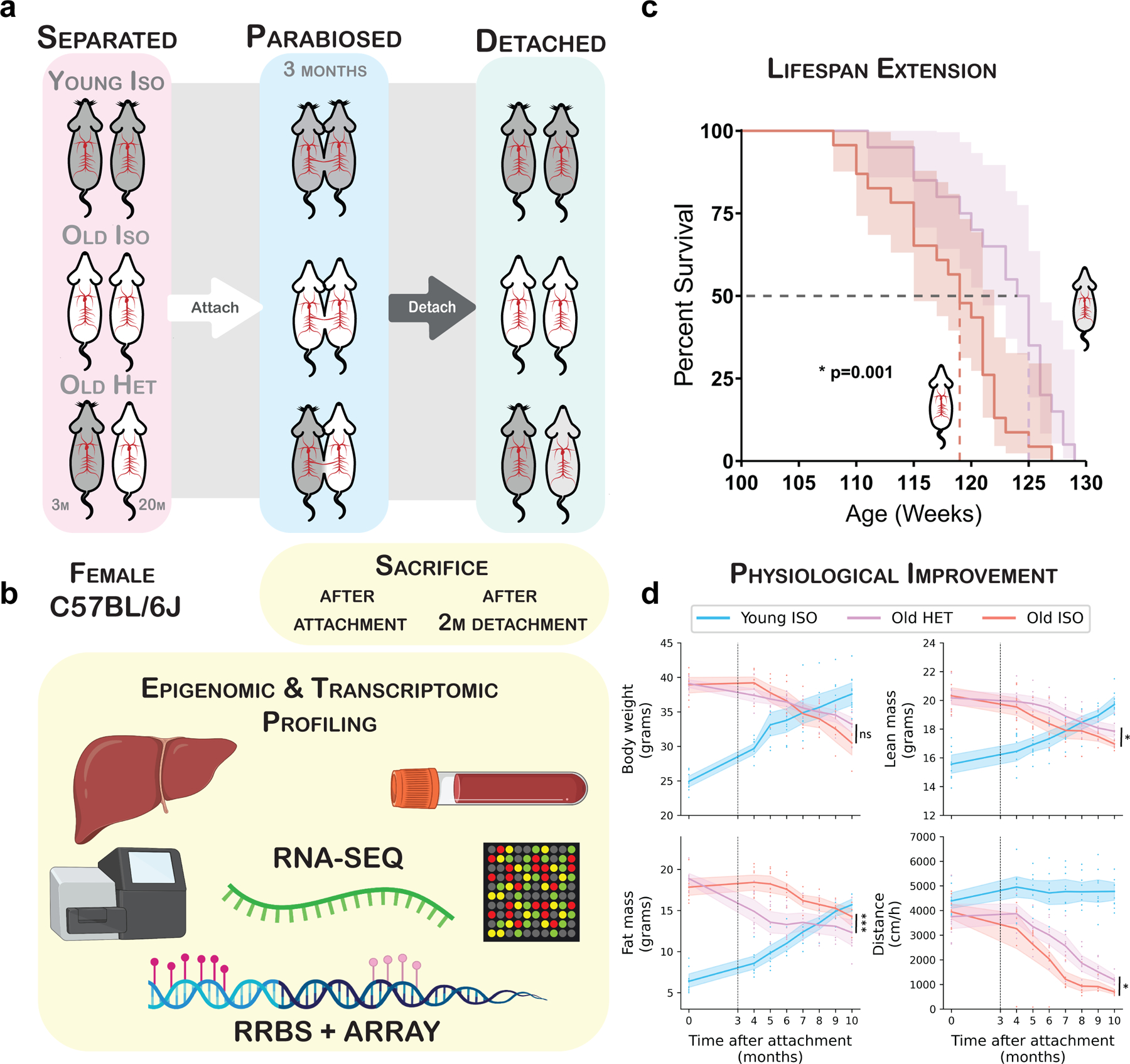
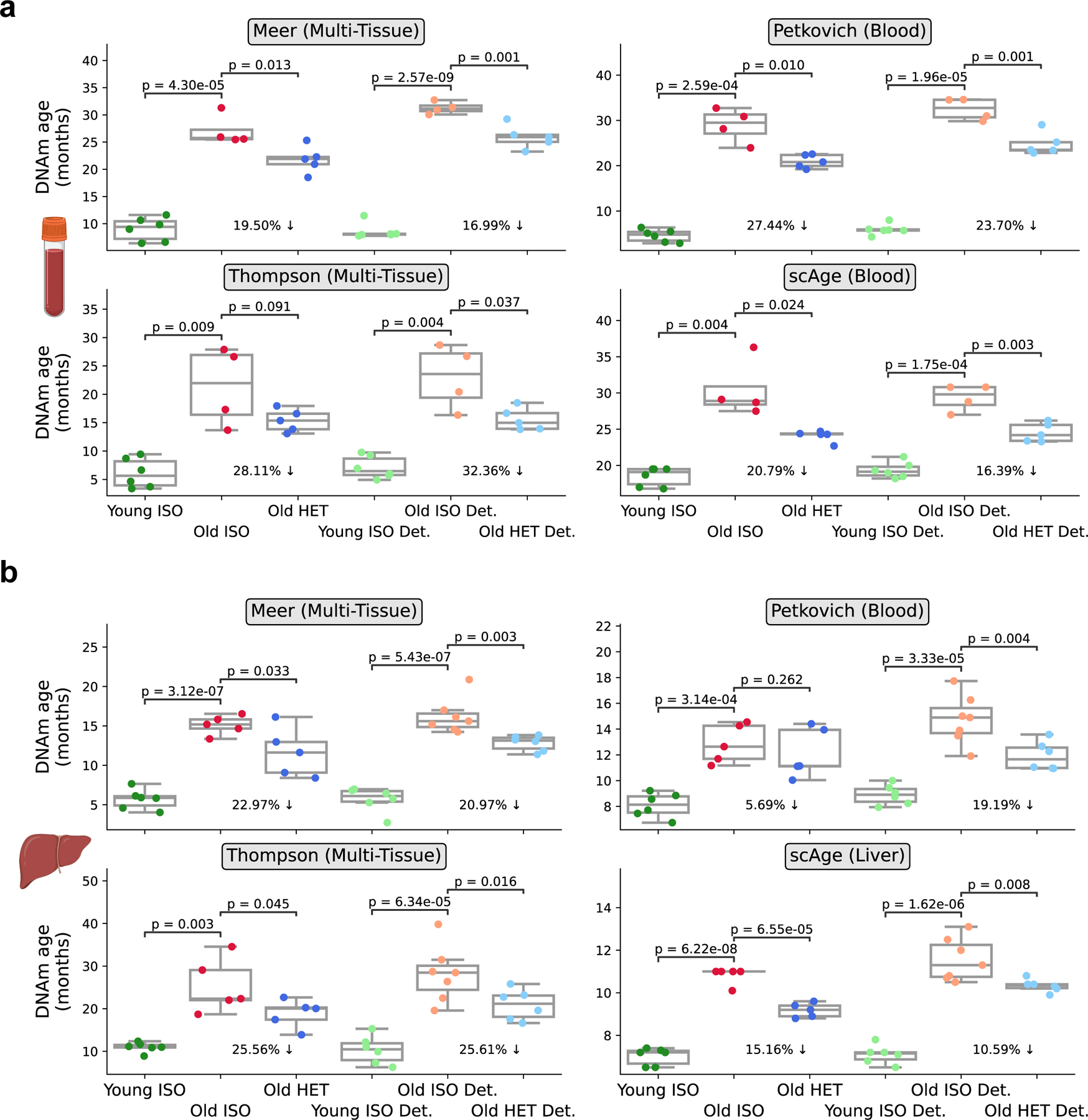
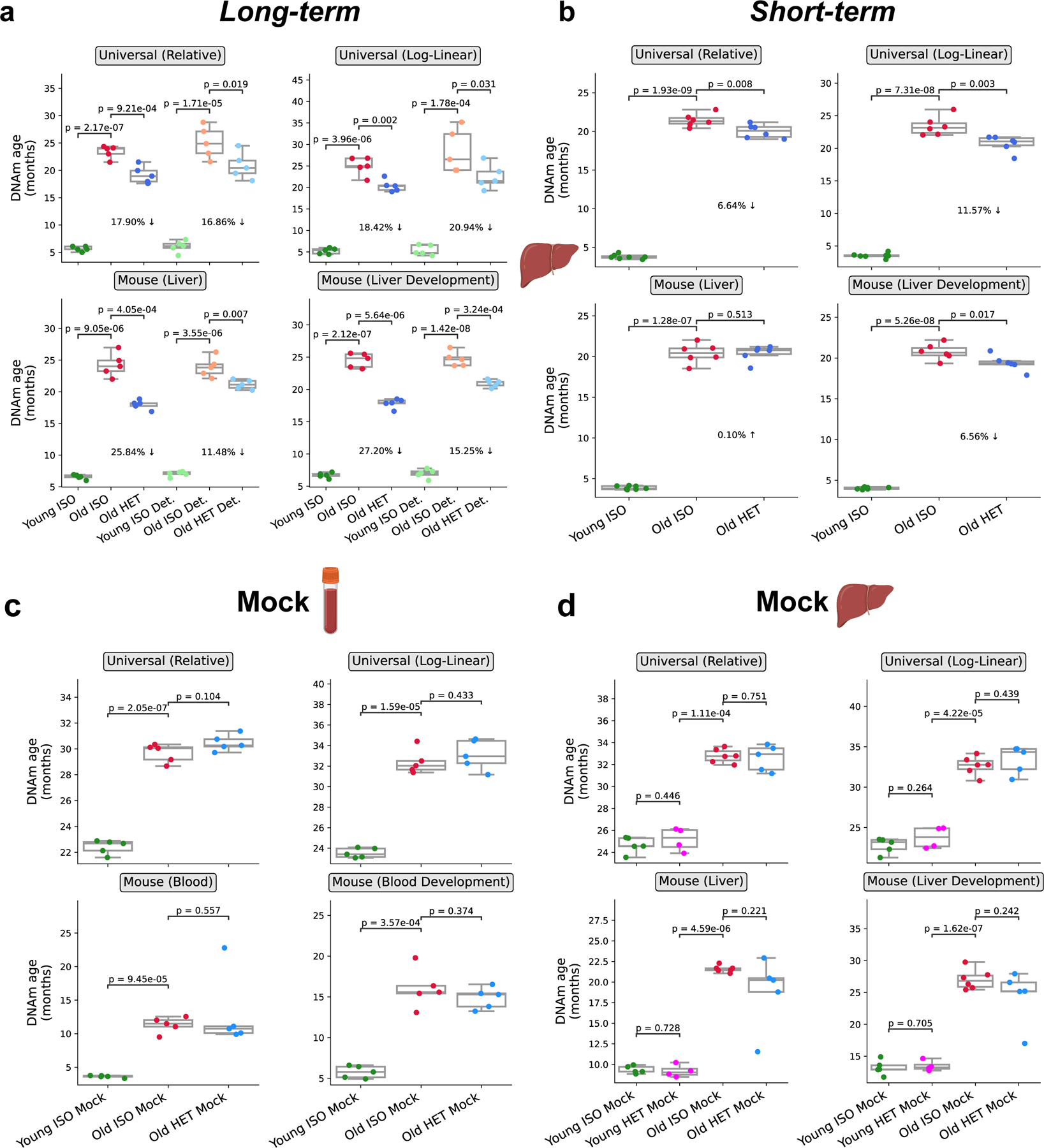
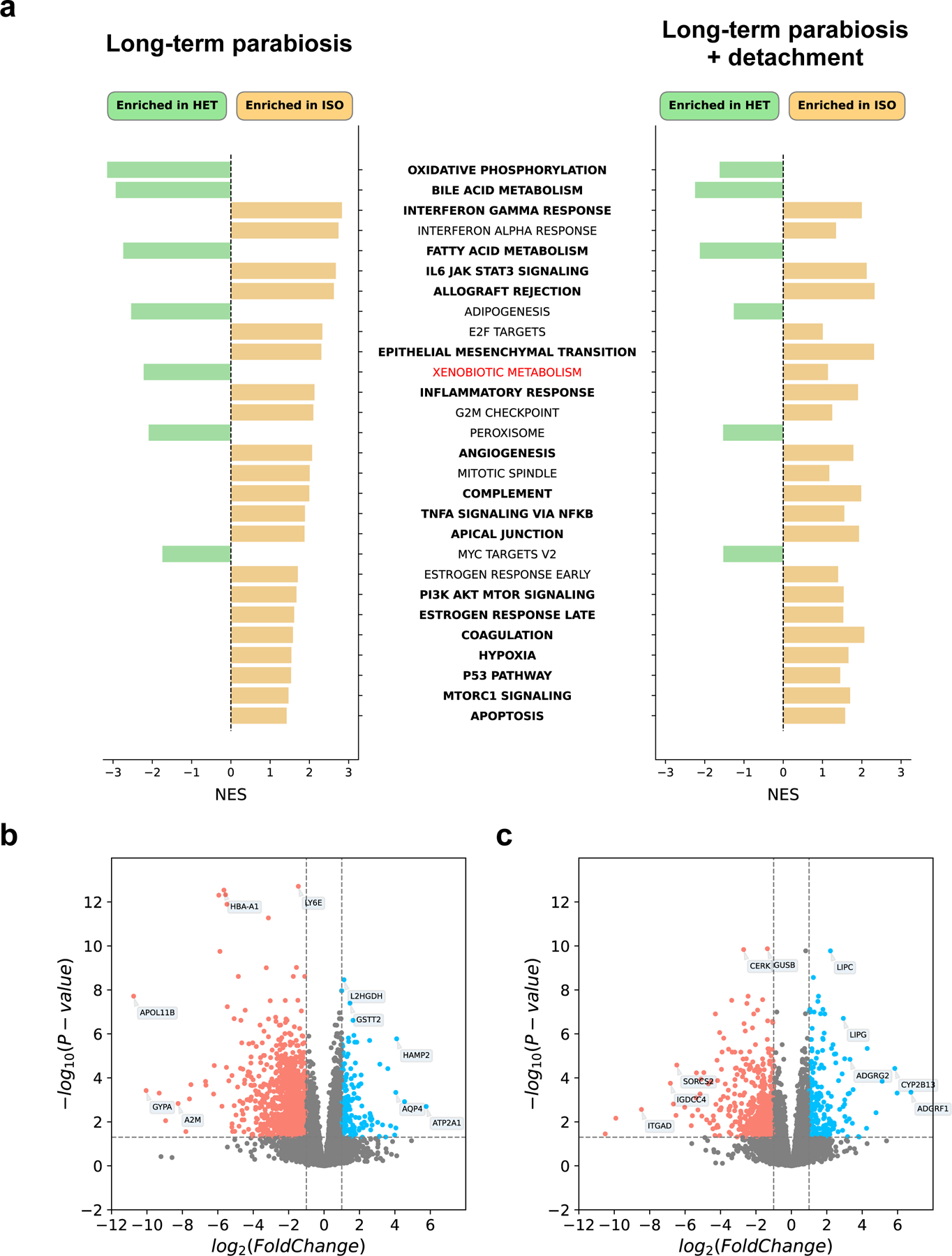
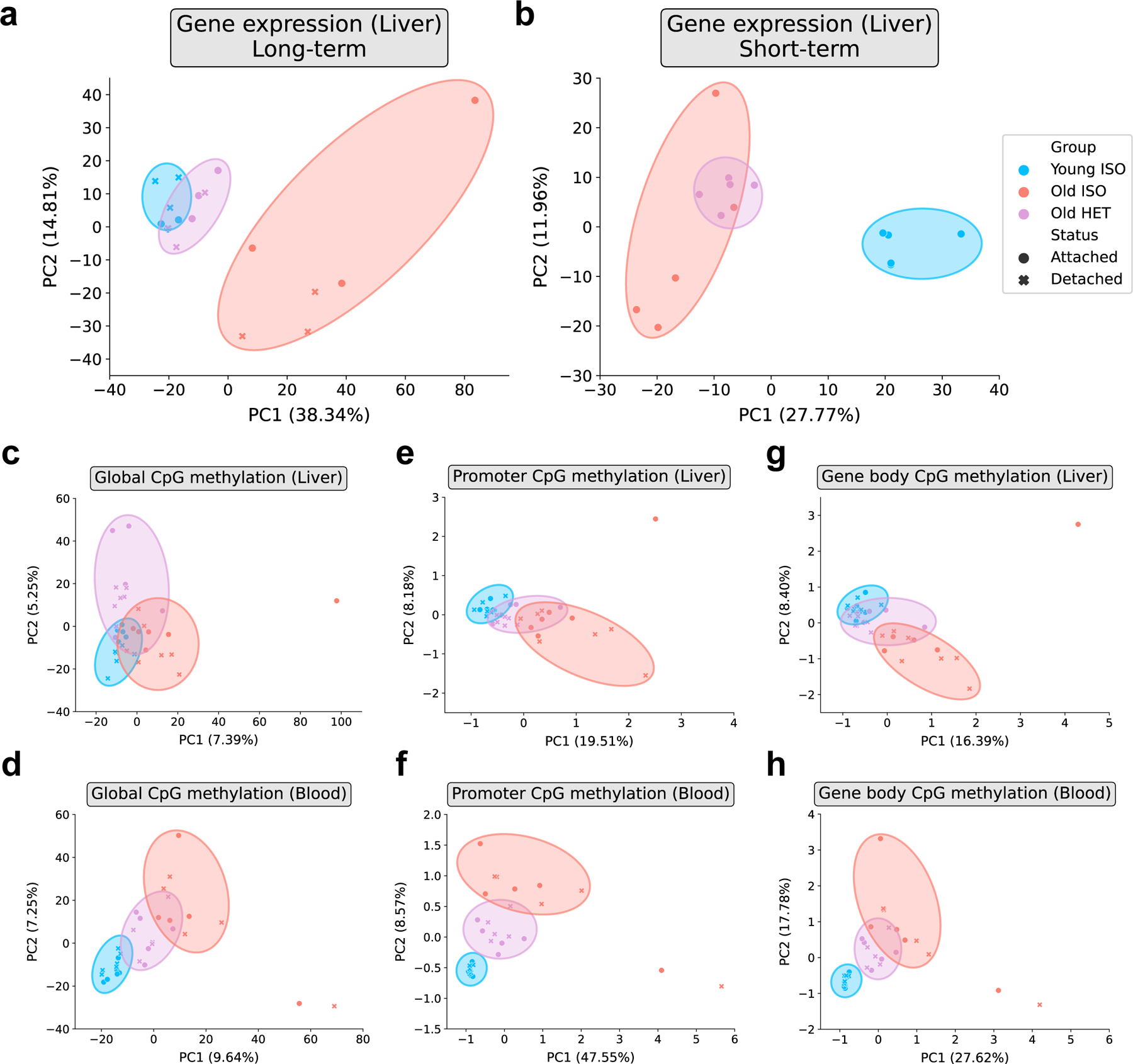
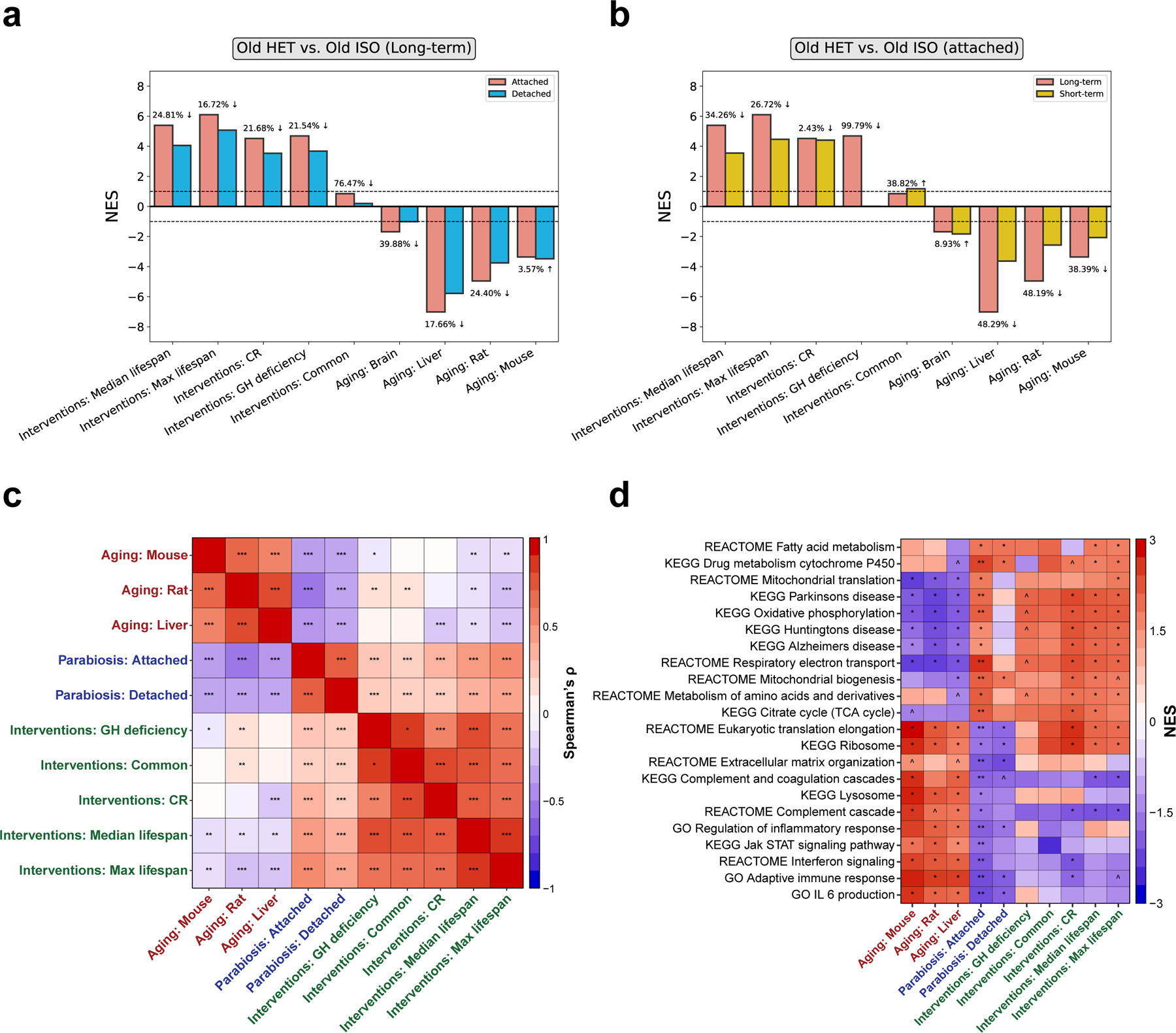
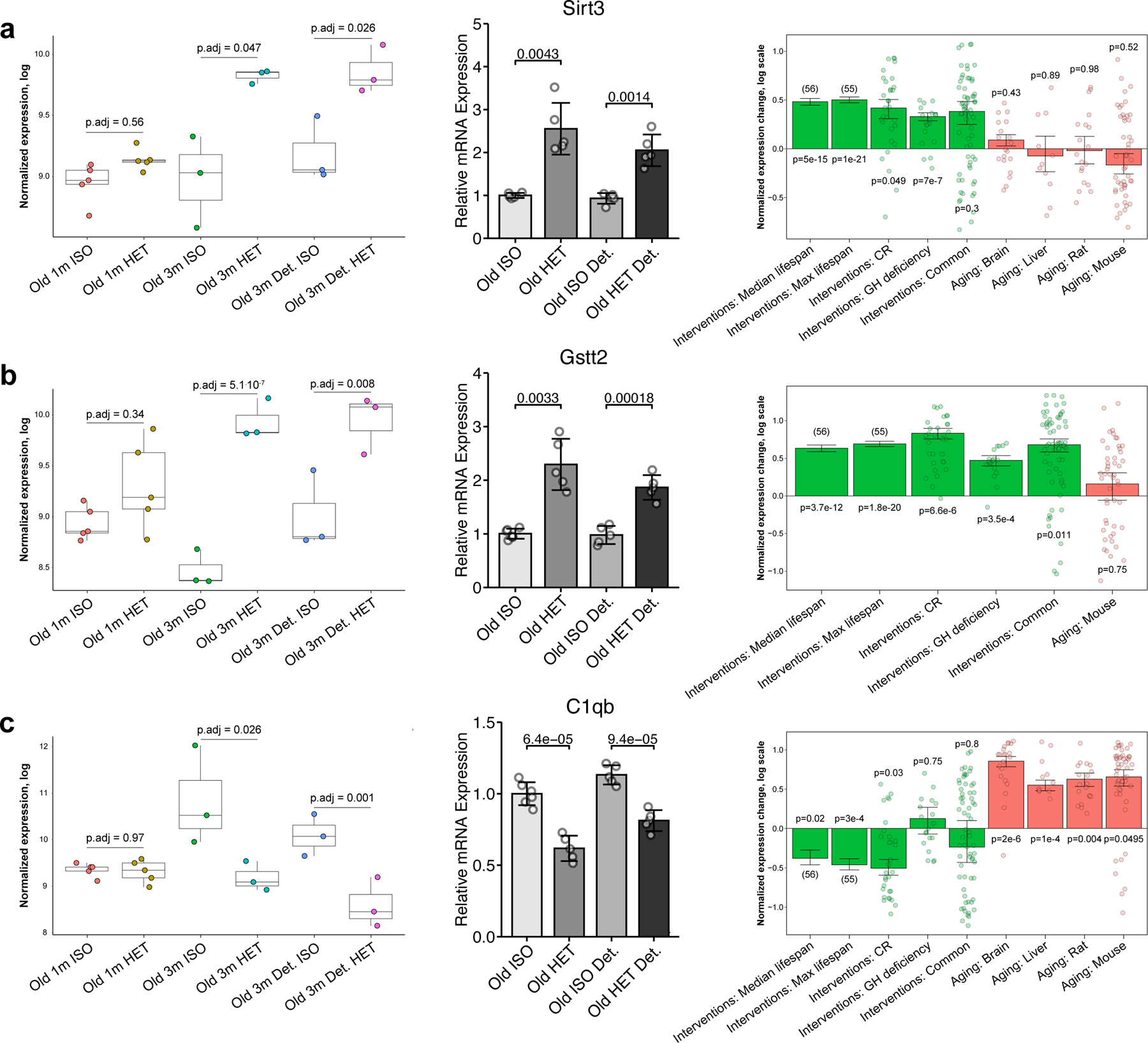
Heterochronic parabiosis (HPB) is known for its functional rejuvenation effects across several mouse tissues. However, its impact on biological age and long-term health is unknown. Here we performed extended (3-month) HPB, followed by a 2-month detachment period of anastomosed pairs. Old detached mice exhibited improved physiological parameters and lived longer than control isochronic mice. HPB drastically reduced the epigenetic age of blood and liver based on several clock models using two independent platforms. Remarkably, this rejuvenation effect persisted even after 2 months of detachment. Transcriptomic and epigenomic profiles of anastomosed mice showed an intermediate phenotype between old and young, suggesting a global multi-omic rejuvenation effect. In addition, old HPB mice showed gene expression changes opposite to aging but akin to several life span-extending interventions. Altogether, we reveal that long-term HPB results in lasting epigenetic and transcriptome remodeling, culminating in the extension of life span and health span.
© 2023. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing interests
The authors do not report any conflicts of interest.
Figures

References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
