

Diagnostic yield and novel candidate genes for neurodevelopmental disorders by exome sequencing in an unselected cohort with microcephaly
- PMID: 37501076
- PMCID: PMC10373276
- DOI: 10.1186/s12864-023-09505-z
Diagnostic yield and novel candidate genes for neurodevelopmental disorders by exome sequencing in an unselected cohort with microcephaly
Abstract
Objectives: Microcephaly is caused by reduced brain volume and most usually associated with a variety of neurodevelopmental disorders (NDDs). To provide an overview of the diagnostic yield of whole exome sequencing (WES) and promote novel candidates in genetically unsolved families, we studied the clinical and genetic landscape of an unselected Chinese cohort of patients with microcephaly.
Methods: We performed WES in an unselected cohort of 103 NDDs patients with microcephaly as one of the features. Full evaluation of potential novel candidate genes was applied in genetically undiagnosed families. Functional validations of selected variants were conducted in cultured cells. To augment the discovery of novel candidates, we queried our genomic sequencing data repository for additional likely disease-causing variants in the identified candidate genes.
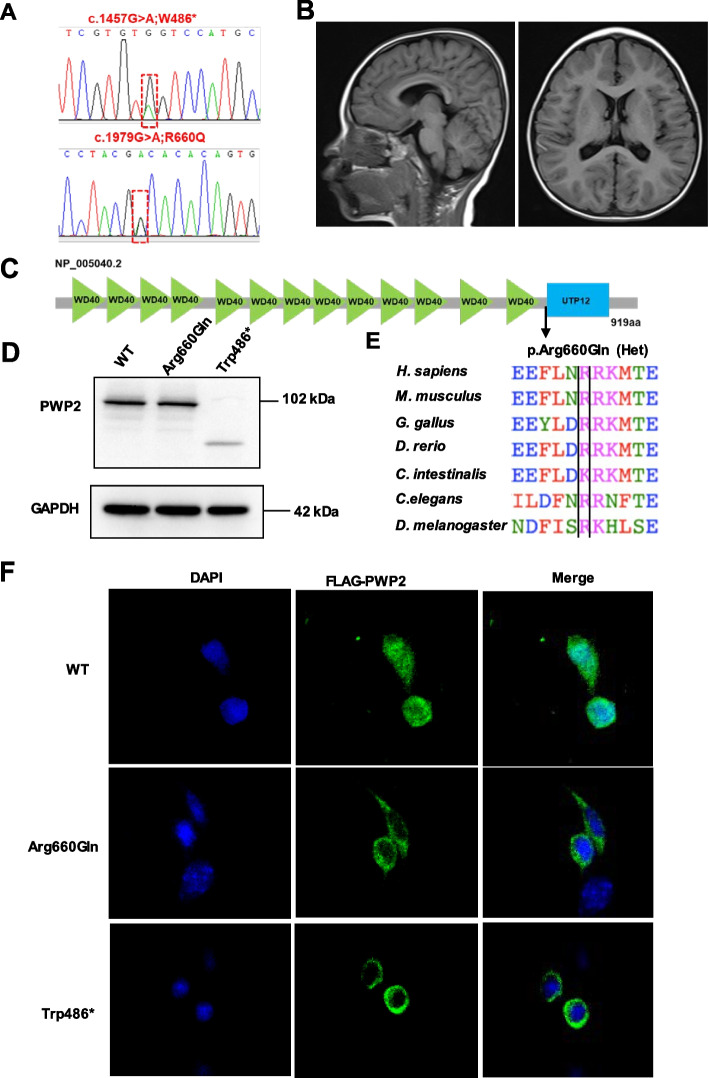
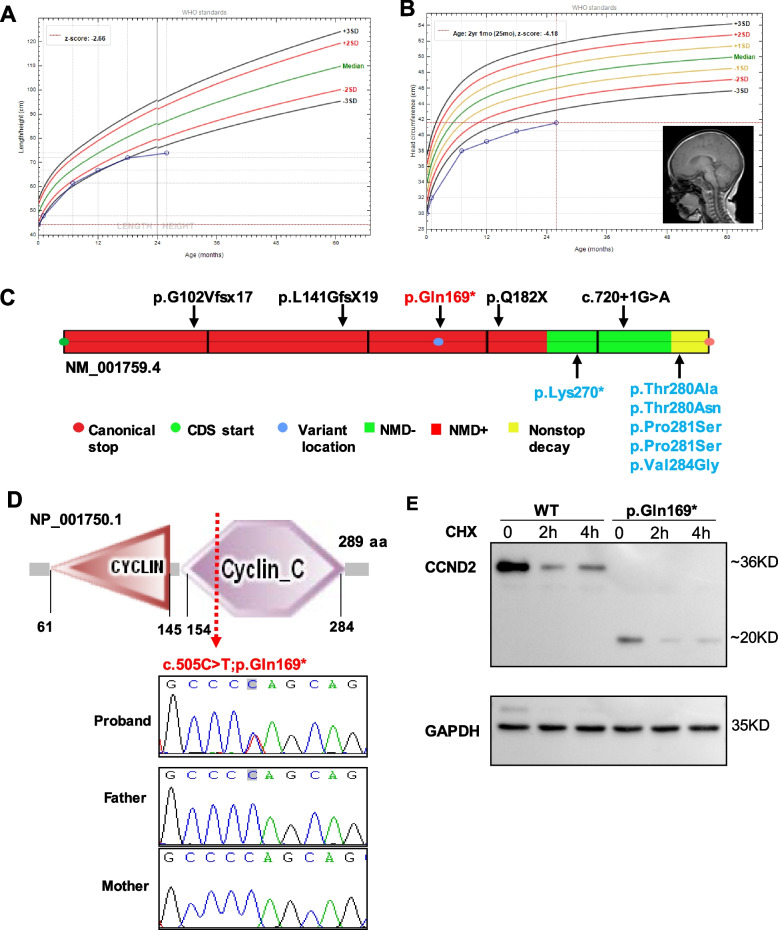
Results: In 65 families (63.1%), causative sequence variants (SVs) and clinically relevant copy number variants (CNVs) with a pathogenic or likely pathogenic (P/LP) level were identified. By incorporating coverage analysis to WES, a pathogenic or likely pathogenic CNV was detected in 15 families (16/103, 15.5%). In another eight families (8/103, 7.8%), we identified variants in newly reported gene (CCND2) and potential novel neurodevelopmental disorders /microcephaly candidate genes, which involved in cell cycle and division (PWP2, CCND2), CDC42/RAC signaling related actin cytoskeletal organization (DOCK9, RHOF), neurogenesis (ELAVL3, PPP1R9B, KCNH3) and transcription regulation (IRF2BP1). By looking into our data repository of 5066 families with NDDs, we identified additional two cases with variants in DOCK9 and PPP1R9B, respectively.
Conclusion: Our results expand the morbid genome of monogenic neurodevelopmental disorders and support the adoption of WES as a first-tier test for individuals with microcephaly.
Keywords: Microcephaly; Neurodevelopmental disorders; Novel candidates; Whole exome sequencing.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures



References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
