

This is a preprint.
Integrated single-cell multiomics uncovers foundational regulatory mechanisms of lens development and pathology
- PMID: 37502967
- PMCID: PMC10369908
- DOI: 10.1101/2023.07.10.548451
Integrated single-cell multiomics uncovers foundational regulatory mechanisms of lens development and pathology
Update in
-
Integrated single-cell multiomics uncovers foundational regulatory mechanisms of lens development and pathology.Development. 2024 Jan 1;151(1):dev202249. doi: 10.1242/dev.202249. Epub 2024 Jan 5. Development. 2024. PMID: 38180241 Free PMC article.
Abstract
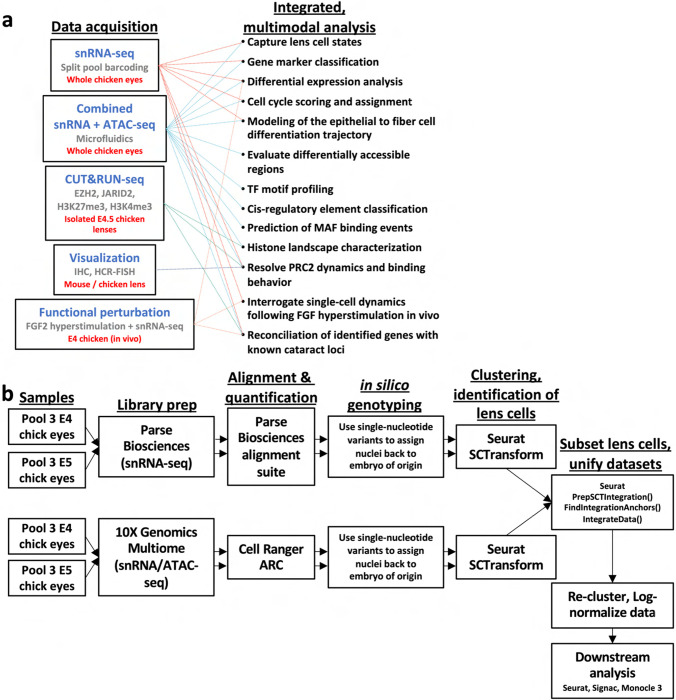
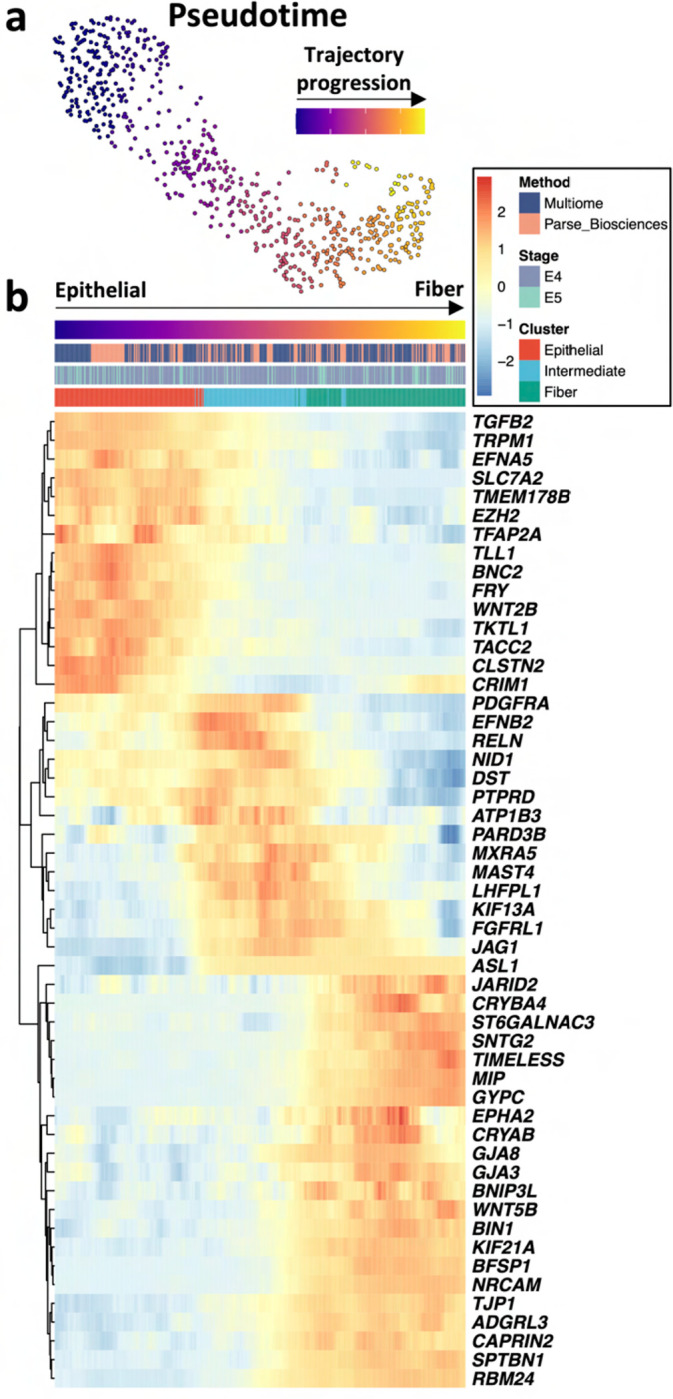
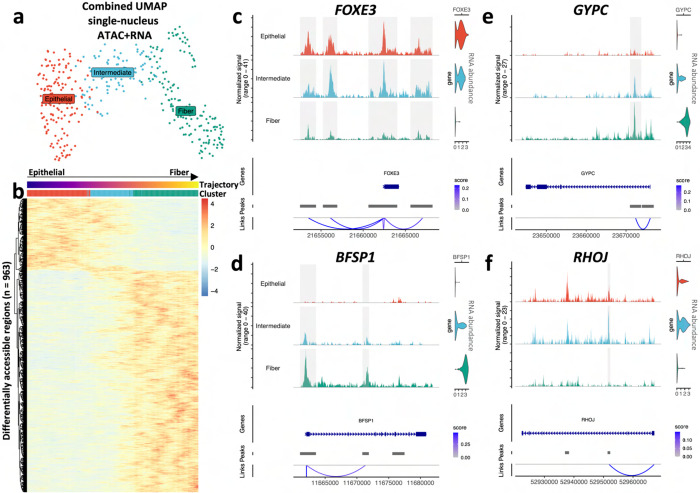
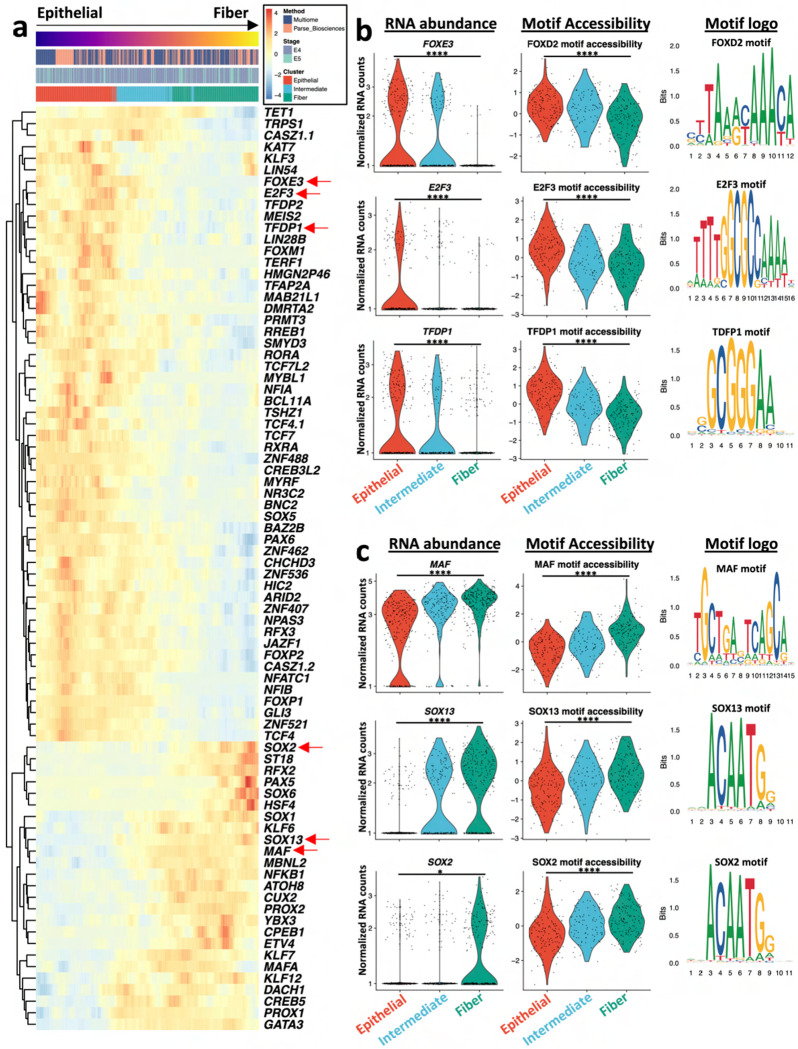
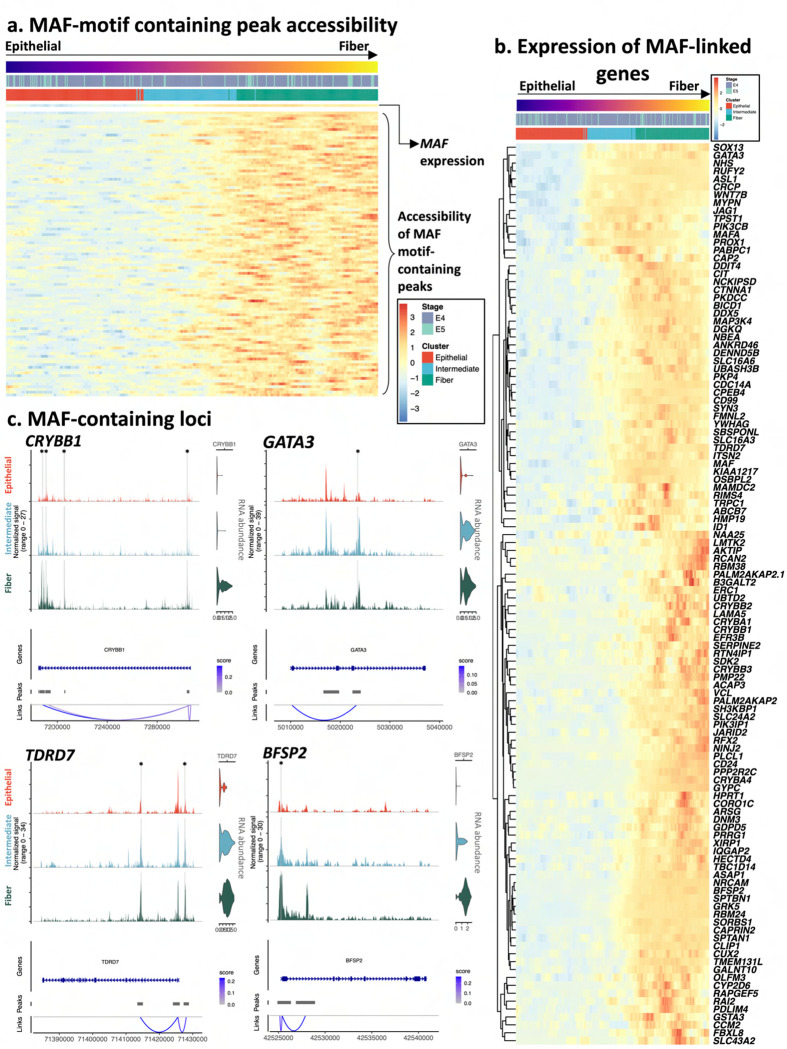
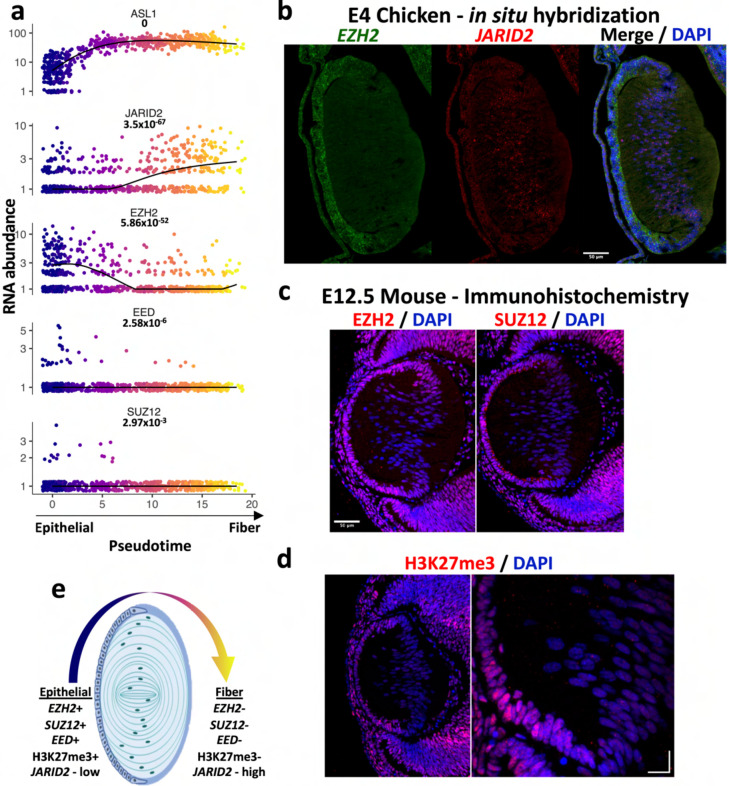
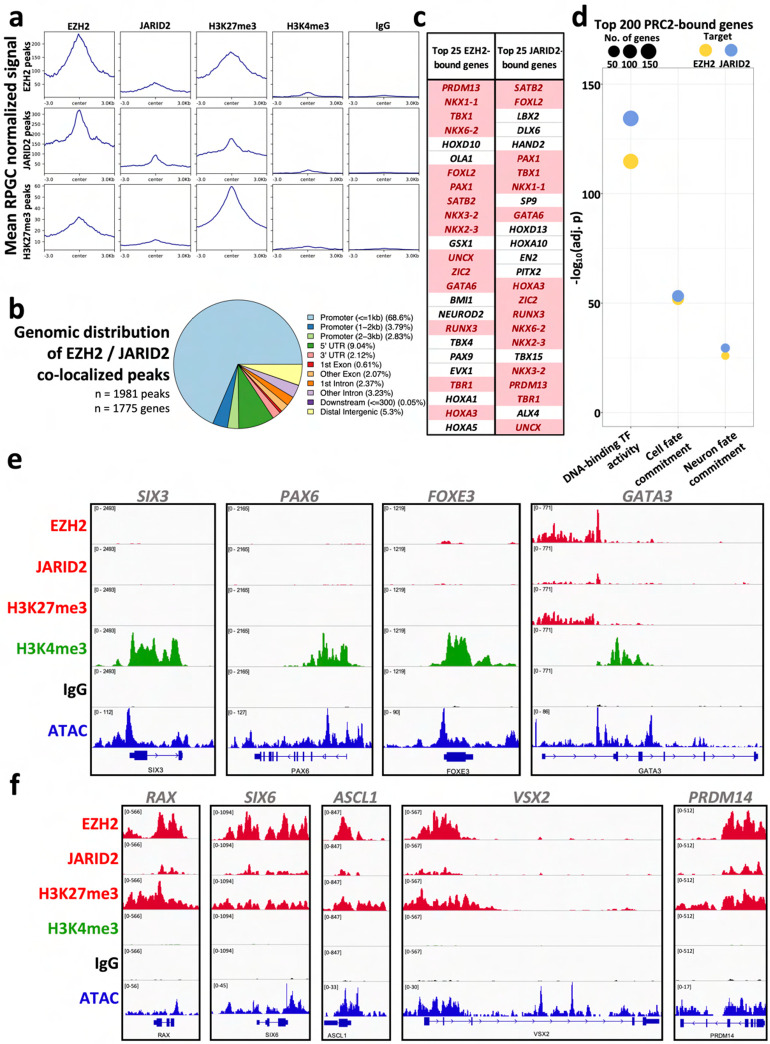
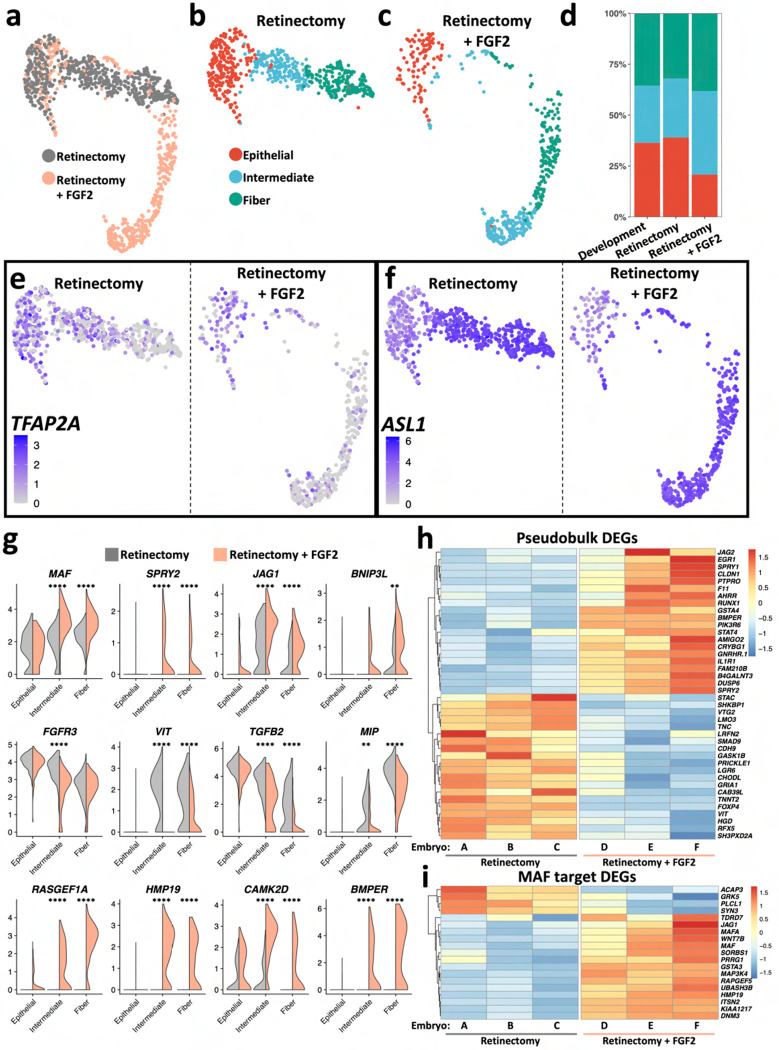
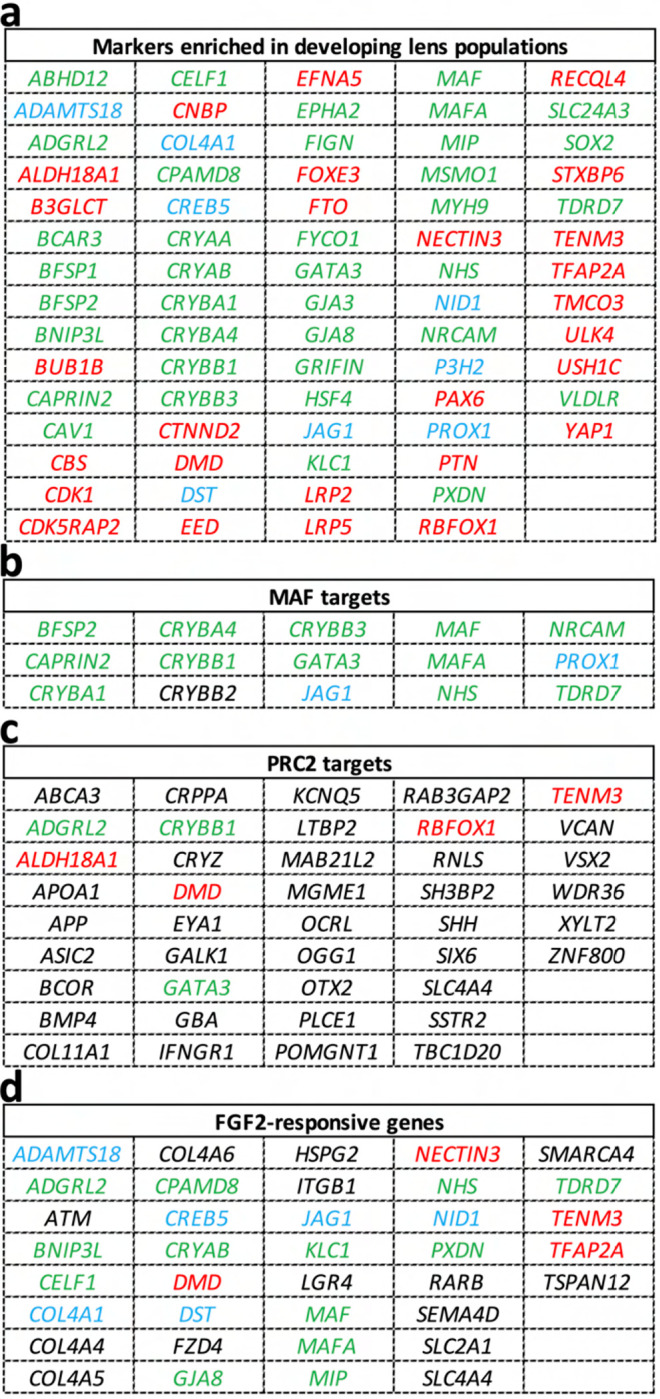
Ocular lens development entails epithelial to fiber cell differentiation, defects in which cause congenital cataract. We report the first single-cell multiomic atlas of lens development, leveraging snRNA-seq, snATAC-seq, and CUT&RUN-seq to discover novel mechanisms of cell fate determination and cataract-linked regulatory networks. A comprehensive profile of cis- and trans-regulatory interactions, including for the cataract-linked transcription factor MAF, is established across a temporal trajectory of fiber cell differentiation. Further, we divulge a conserved epigenetic paradigm of cellular differentiation, defined by progressive loss of H3K27 methylation writer Polycomb repressive complex 2 (PRC2). PRC2 localizes to heterochromatin domains across master-regulator transcription factor gene bodies, suggesting it safeguards epithelial cell fate. Moreover, we demonstrate that FGF hyper-stimulation in vivo leads to MAF network activation and the emergence of novel lens cell states. Collectively, these data depict a comprehensive portrait of lens fiber cell differentiation, while defining regulatory effectors of cell identity and cataract formation.
Conflict of interest statement
Competing Interests The authors declare no competing interests.
Figures

References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources