

This is a preprint.
Patterns of recombination in snakes reveal a tug of war between PRDM9 and promoter-like features
- PMID: 37502971
- PMCID: PMC10369914
- DOI: 10.1101/2023.07.11.548536
Patterns of recombination in snakes reveal a tug of war between PRDM9 and promoter-like features
Update in
-
Patterns of recombination in snakes reveal a tug-of-war between PRDM9 and promoter-like features.Science. 2024 Feb 23;383(6685):eadj7026. doi: 10.1126/science.adj7026. Epub 2024 Feb 23. Science. 2024. PMID: 38386752
Abstract
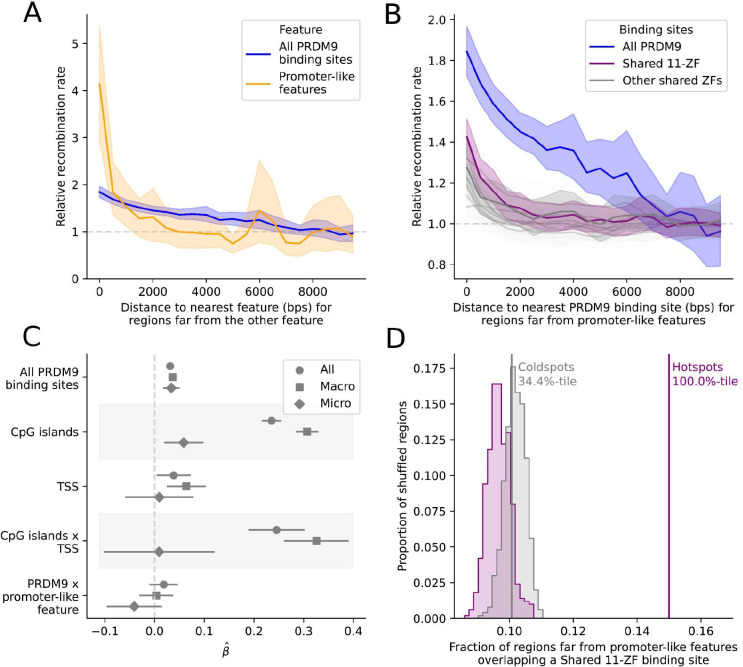
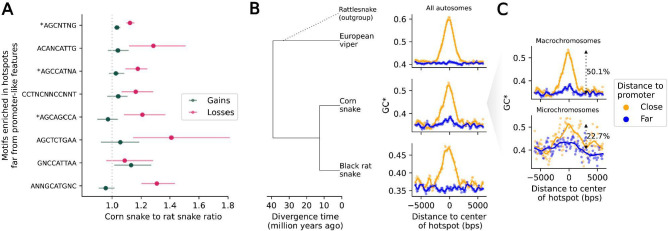
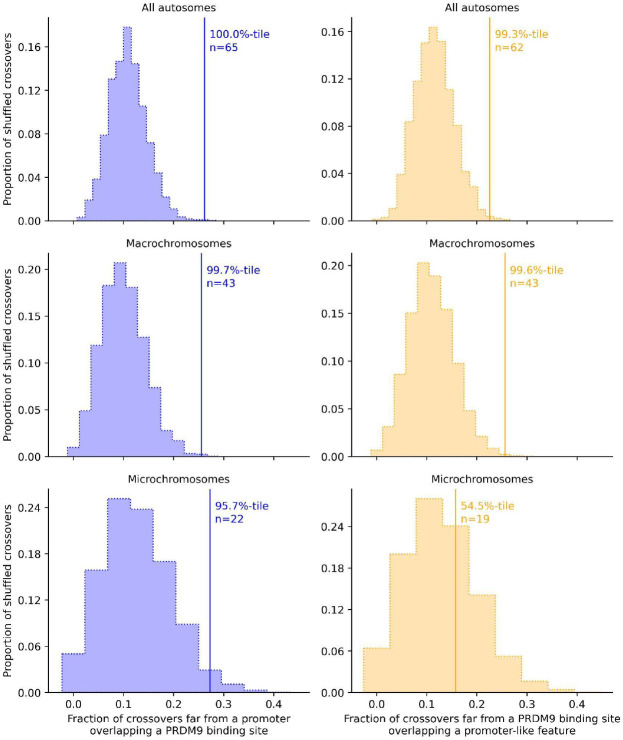
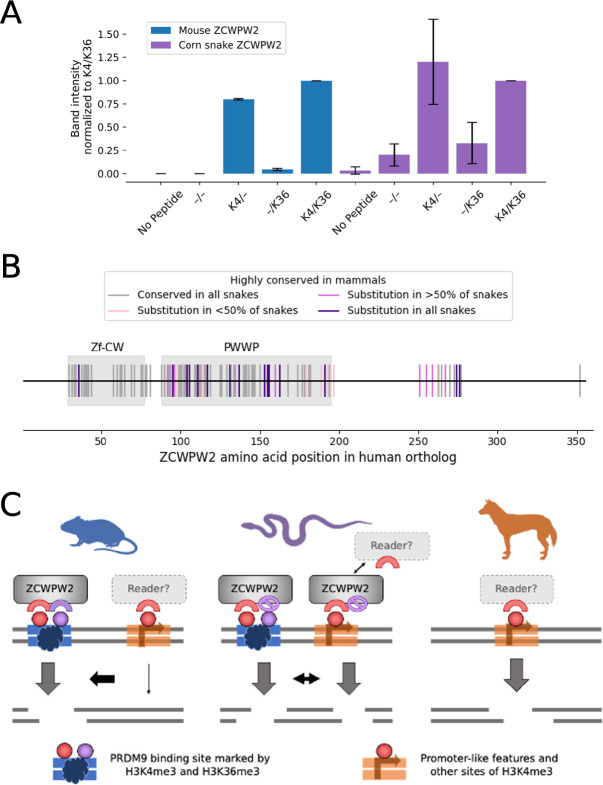
In vertebrates, there are two known mechanisms by which meiotic recombination is directed to the genome: in humans, mice, and other mammals, recombination occurs almost exclusively where the protein PRDM9 binds, while in species lacking an intact PRDM9, such as birds and canids, recombination rates are elevated near promoter-like features. To test if PRDM9 also directs recombination in non-mammalian vertebrates, we focused on an exemplar species, the corn snake (Pantherophis guttatus). Unlike birds, this species possesses a single, intact PRDM9 ortholog. By inferring historical recombination rates along the genome from patterns of linkage disequilibrium and identifying crossovers in pedigrees, we found that PRDM9 specifies the location of recombination events outside of mammals. However, we also detected an independent effect of promoter-like features on recombination, which is more pronounced on macro- than microchromosomes. Thus, our findings reveal that the uses of PRDM9 and promoter-like features are not mutually-exclusive, and instead reflect a tug of war, which varies in strength along the genome and is more lopsided in some species than others.
Conflict of interest statement
Competing interests: Authors declare that they have no competing interests.
Figures

References
-
- de Massy B., Initiation of meiotic recombination: how and where? Conservation and specificities among eukaryotes. Annu. Rev. Genet. 47, 563–599 (2013). - PubMed
-
- Zhou Y., Shen B., Jiang J., Padhi A., Park K.-E., Oswalt A., Sattler C. G., Telugu B. P., Chen H., Cole J. B., Liu G. E., Ma L., Construction of PRDM9 allele-specific recombination maps in cattle using large-scale pedigree analysis and genome-wide single sperm genomics. DNA Res. 25, 183–194 (2018). - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources