

This is a preprint.
The genetic diversity of Nipah virus across spatial scales
- PMID: 37502973
- PMCID: PMC10370237
- DOI: 10.1101/2023.07.14.23292668
The genetic diversity of Nipah virus across spatial scales
Update in
-
The Genetic Diversity of Nipah Virus Across Spatial Scales.J Infect Dis. 2024 Dec 16;230(6):e1235-e1244. doi: 10.1093/infdis/jiae221. J Infect Dis. 2024. PMID: 38682164 Free PMC article.
Abstract
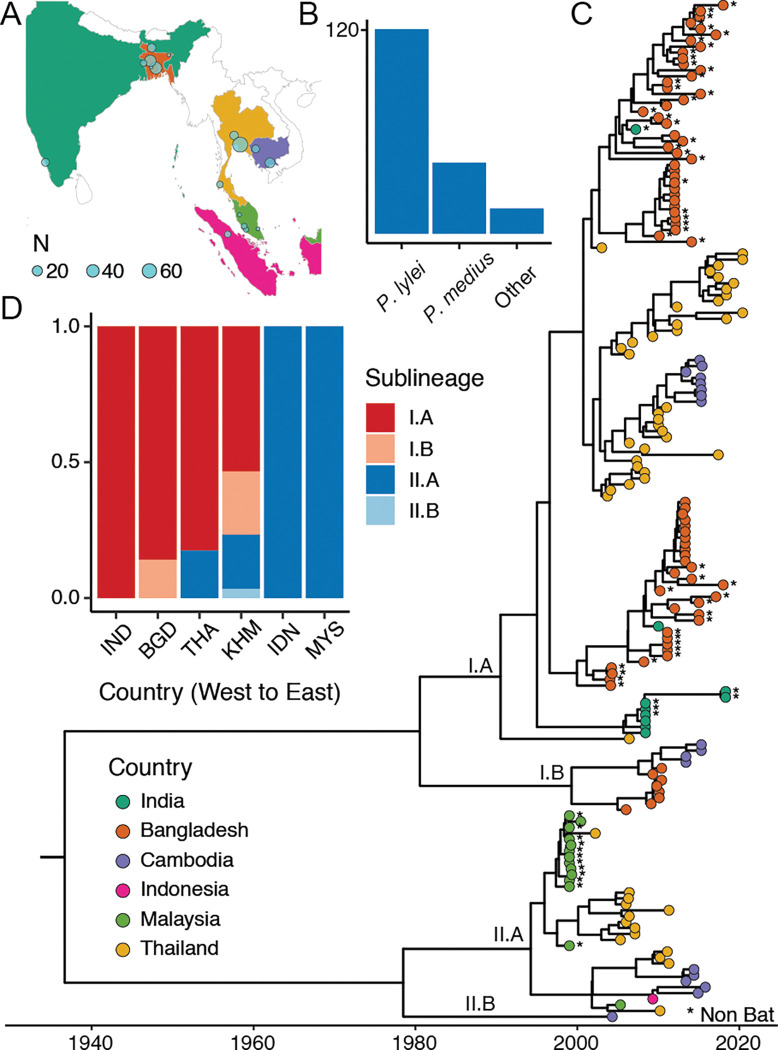
Nipah virus (NiV), a highly lethal virus in humans, circulates silently in Pteropus bats throughout South and Southeast Asia. Difficulty in obtaining genomes from bats means we have a poor understanding of NiV diversity, including how many lineages circulate within a roost and the spread of NiV over increasing spatial scales. Here we develop phylogenetic approaches applied to the most comprehensive collection of genomes to date (N=257, 175 from bats, 73 from humans) from six countries over 22 years (1999-2020). In Bangladesh, where most human infections occur, we find evidence of increased spillover risk from one of the two co-circulating sublineages. We divide the four major NiV sublineages into 15 genetic clusters (emerged 20-44 years ago). Within any bat roost, there are an average of 2.4 co-circulating genetic clusters, rising to 5.5 clusters at areas of 1,500-2,000 km2. Using Approximate Bayesian Computation fit to a spatial signature of viral diversity, we estimate that each genetic cluster occupies an average area of 1.3 million km2 (95%CI: 0.6-2.3 million), with 14 clusters in an area of 100,000 km2 (95%CI: 6-24). In the few sites in Bangladesh and Cambodia where genomic surveillance has been concentrated, we estimate that most of the genetic clusters have been identified, but only ~15% of overall NiV diversity has been uncovered. Our findings are consistent with entrenched co-circulation of distinct lineages, even within individual roosts, coupled with slow migration over larger spatial scales.
Figures

References
-
- World Health Organization, NIPAH RESEARCH AND DEVELOPMENT (R&D) ROADMAP (2019) (available at https://cdn.who.int/media/docs/default-source/blue-print/nipah_rdbluepri...)
-
- Nikolay B., Salje H., Hossain M. J., Khan A. K. M. D., Sazzad H. M. S., Rahman M., Daszak P., Ströher U., Pulliam J. R. C., Kilpatrick A. M., Nichol S. T., Klena J. D., Sultana S., Afroj S., Luby S. P., Cauchemez S., Gurley E. S., Transmission of Nipah Virus - 14 Years of Investigations in Bangladesh. N. Engl. J. Med. 380, 1804–1814 (2019). - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources