

This is a preprint.
Contextual AI models for single-cell protein biology
- PMID: 37503080
- PMCID: PMC10370131
- DOI: 10.1101/2023.07.18.549602
Contextual AI models for single-cell protein biology
Update in
-
Contextual AI models for single-cell protein biology.Nat Methods. 2024 Aug;21(8):1546-1557. doi: 10.1038/s41592-024-02341-3. Epub 2024 Jul 22. Nat Methods. 2024. PMID: 39039335 Free PMC article.
Abstract
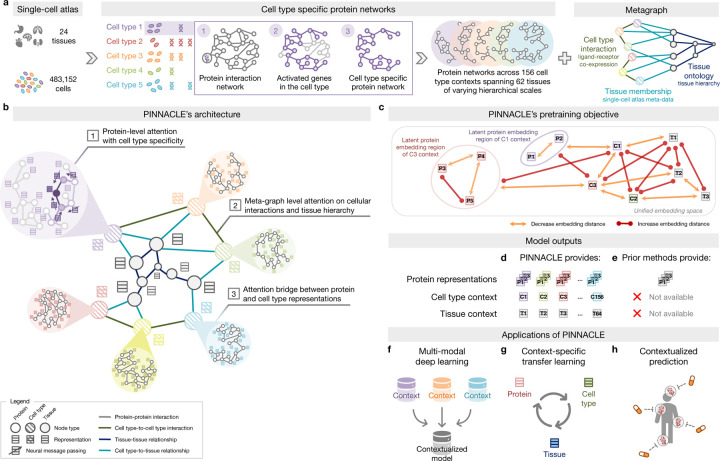
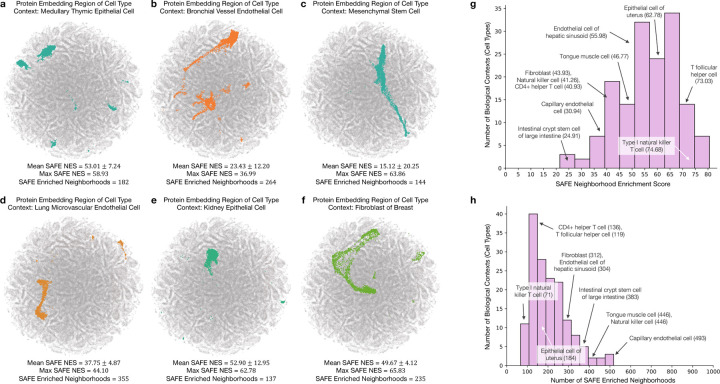
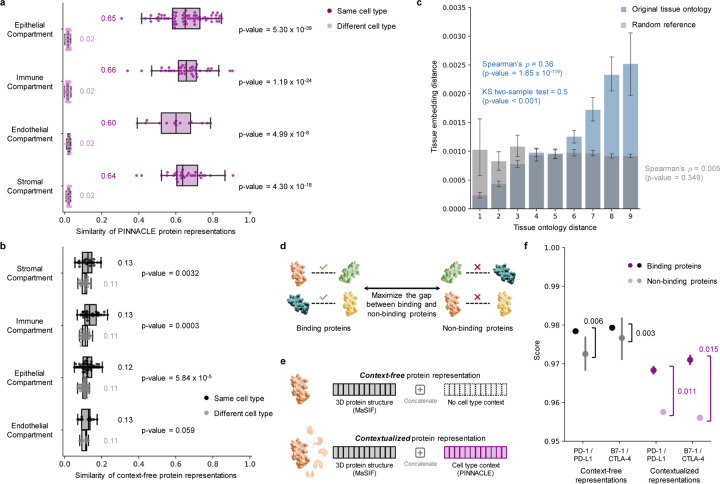
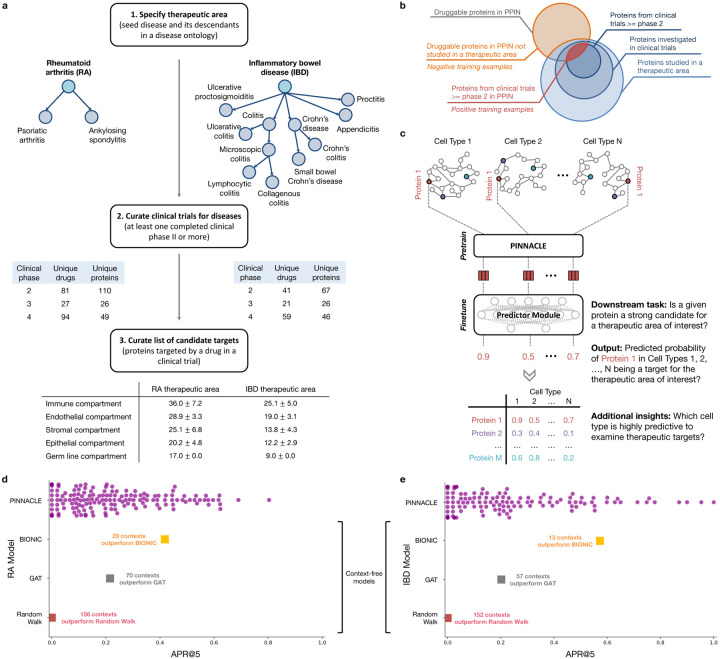
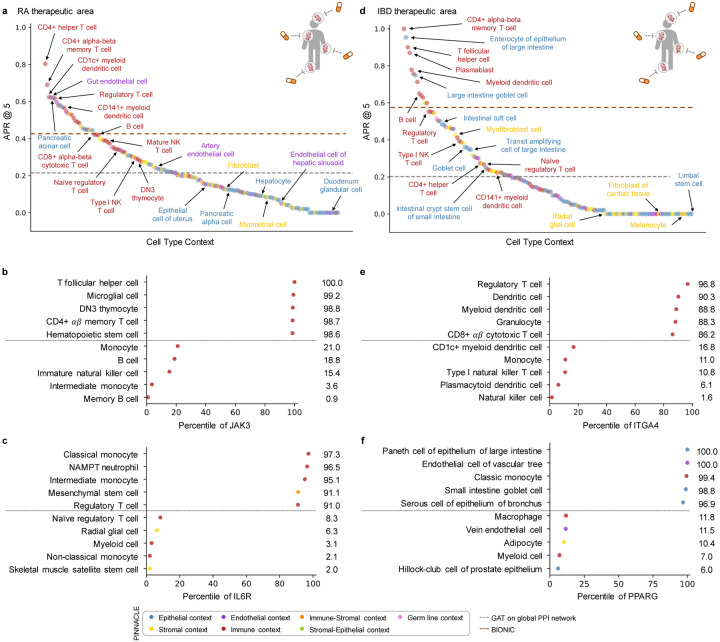
Understanding protein function and developing molecular therapies require deciphering the cell types in which proteins act as well as the interactions between proteins. However, modeling protein interactions across biological contexts remains challenging for existing algorithms. Here, we introduce Pinnacle, a geometric deep learning approach that generates context-aware protein representations. Leveraging a multi-organ single-cell atlas, Pinnacle learns on contextualized protein interaction networks to produce 394,760 protein representations from 156 cell type contexts across 24 tissues. Pinnacle's embedding space reflects cellular and tissue organization, enabling zero-shot retrieval of the tissue hierarchy. Pretrained protein representations can be adapted for downstream tasks: enhancing 3D structure-based representations for resolving immuno-oncological protein interactions, and investigating drugs' effects across cell types. Pinnacle outperforms state-of-the-art models in nominating therapeutic targets for rheumatoid arthritis and inflammatory bowel diseases, and pinpoints cell type contexts with higher predictive capability than context-free models. Pinnacle's ability to adjust its outputs based on the context in which it operates paves way for large-scale context-specific predictions in biology.
Conflict of interest statement
Competing interests. D.M. and A.V. are currently employed by F. Hoffmann-La Roche Ltd. The remaining authors declare no competing interests.
Figures
References
-
- Lund-Johansen F., Tran T. & Mehta A. Towards reproducibility in large-scale analysis of protein–protein interactions. Nature Methods 18, 720–721 (2021). - PubMed
-
- Kustatscher G. et al. Understudied proteins: opportunities and challenges for functional proteomics. Nature Methods 19, 774–779 (2022). - PubMed
-
- Gainza P. et al. Deciphering interaction fingerprints from protein molecular surfaces using geometric deep learning. Nature Methods 17, 184–192 (2019). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources