

This is a preprint.
Integrated genomic and functional analyses of human skin-associated Staphylococcus reveals extensive inter- and intra-species diversity
- PMID: 37503282
- PMCID: PMC10370188
- DOI: 10.1101/2023.06.22.546190
Integrated genomic and functional analyses of human skin-associated Staphylococcus reveals extensive inter- and intra-species diversity
Update in
-
Integrated genomic and functional analyses of human skin-associated Staphylococcus reveal extensive inter- and intra-species diversity.Proc Natl Acad Sci U S A. 2023 Nov 21;120(47):e2310585120. doi: 10.1073/pnas.2310585120. Epub 2023 Nov 13. Proc Natl Acad Sci U S A. 2023. PMID: 37956283 Free PMC article.
Abstract
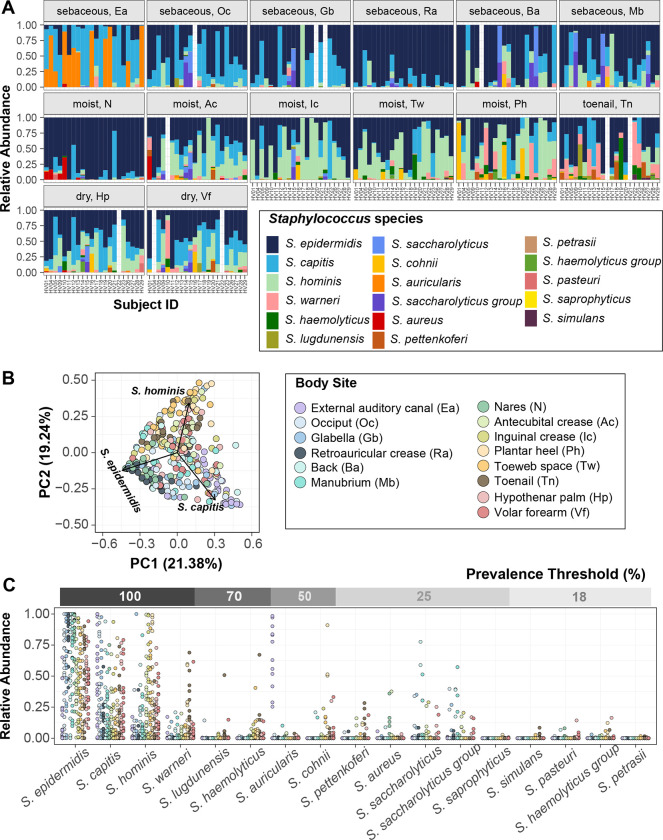
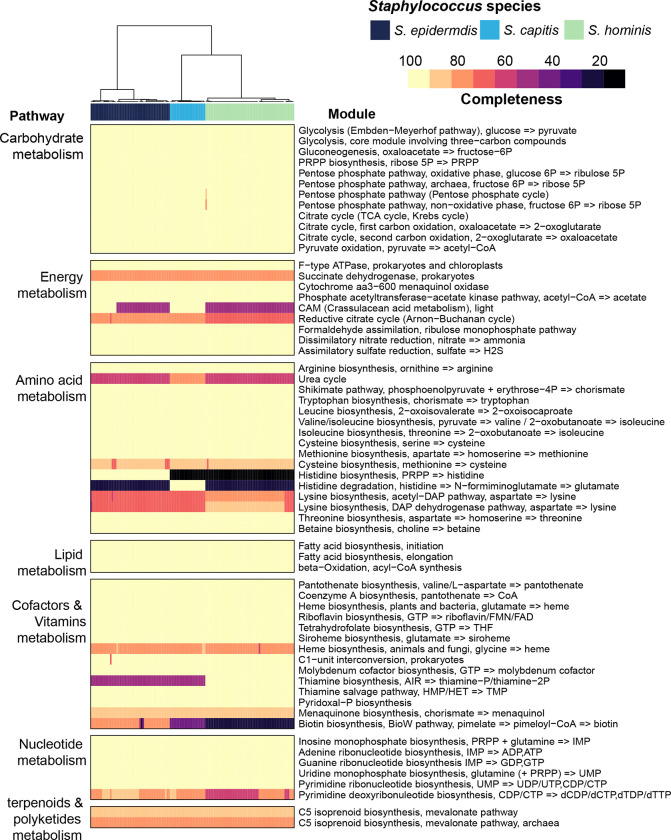
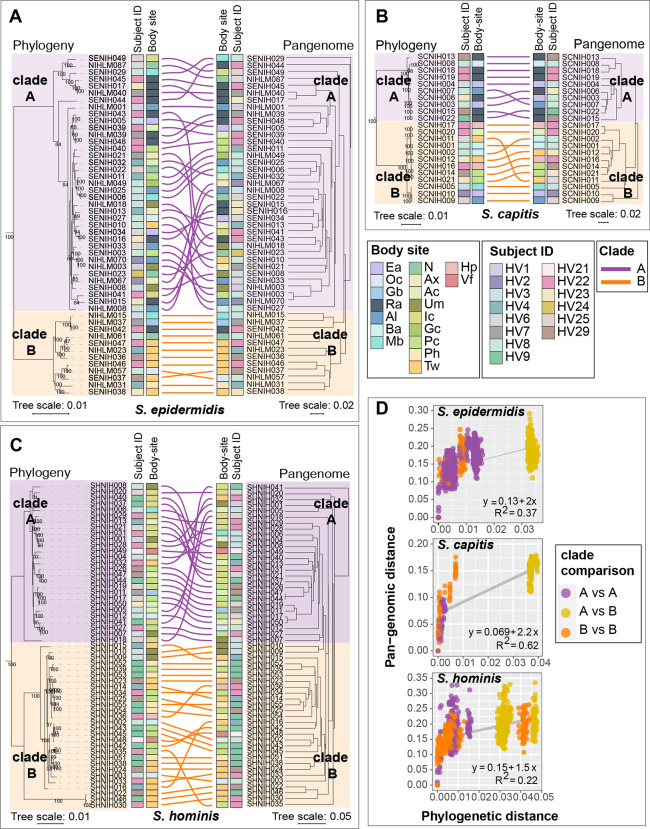
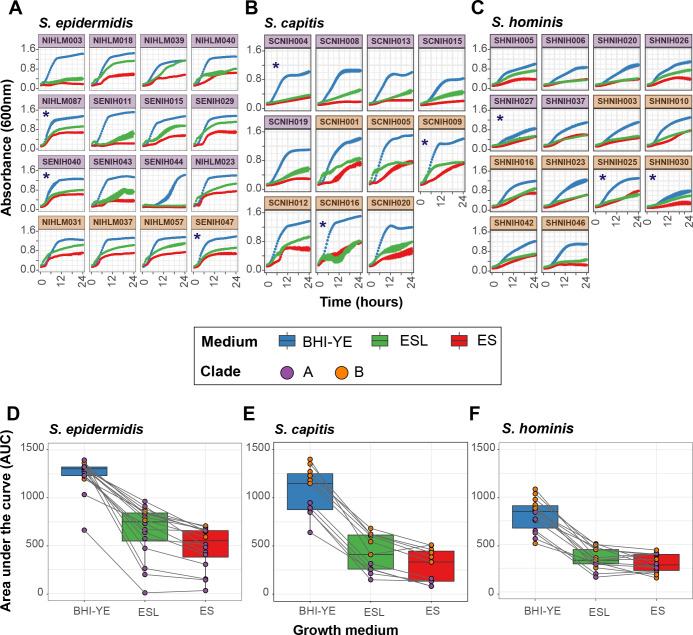
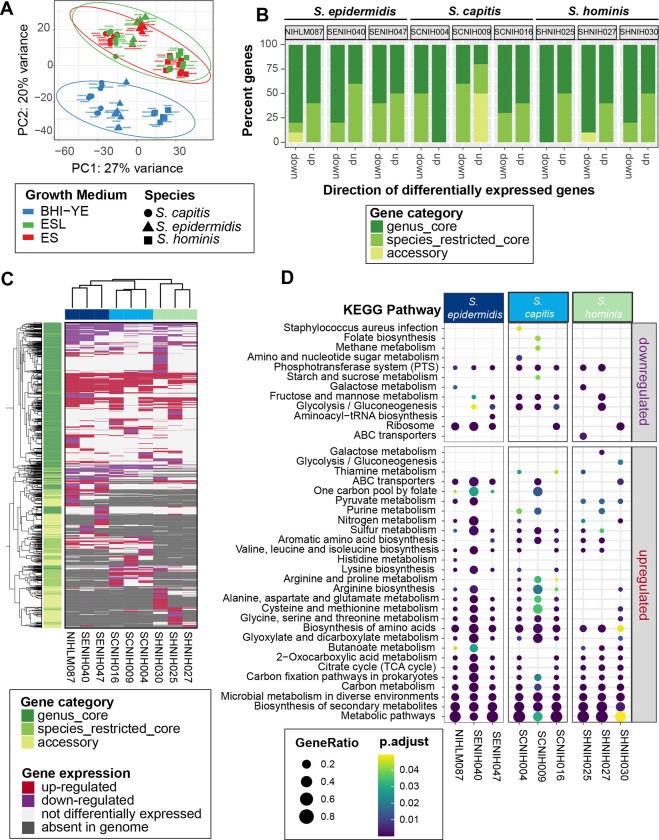
Human skin is stably colonized by a distinct microbiota that functions together with epidermal cells to maintain a protective physical barrier. Staphylococcus, a prominent genus of the skin microbiota, participates in colonization resistance, tissue repair, and host immune regulation in strain specific manners. To unlock the potential of engineering skin microbial communities, we aim to fully characterize the functional diversity of this genus within the context of the skin environment. We conducted metagenome and pan-genome analyses of isolates obtained from distinct body sites of healthy volunteers, providing a detailed biogeographic depiction of staphylococcal species that colonize our skin. S. epidermidis, S. capitis, and S. hominis were the most abundant species present in all volunteers and were detected at all body sites. Pan-genome analysis of these three species revealed that the genus-core was dominated by central metabolism genes. Species-specific core genes were enriched in host colonization functions. The majority (~68%) of genes were detected only in a fraction of isolate genomes, underscoring the immense strain-specific gene diversity. Conspecific genomes grouped into phylogenetic clades, exhibiting body site preference. Each clade was enriched for distinct gene-sets that are potentially involved in site tropism. Finally, we conducted gene expression studies of select isolates showing variable growth phenotypes in skin-like medium. In vitro expression revealed extensive intra- and inter-species gene expression variation, substantially expanding the functional diversification within each species. Our study provides an important resource for future ecological and translational studies to examine the role of shared and strain-specific staphylococcal genes within the skin environment.
Keywords: Staphylococcus capitis; Staphylococcus epidermidis; Staphylococcus hominis; gene expression; growth curve; metagenomics; pan-genomics; phylogenetics; skin microbiota; staphylococci.
Conflict of interest statement
DECLARATION OF INTERESTS The authors declare no competing interests.
Figures


References
-
- Proksch E., Brandner J. M., Jensen J. M., The skin: an indispensable barrier. Exp Dermatol 17, 1063–1072 (2008). - PubMed
-
- Harris-Tryon T. A., Grice E. A., Microbiota and maintenance of skin barrier function. Science 376, 940–945 (2022). - PubMed
-
- Otto M., Staphylococci in the human microbiome: the role of host and interbacterial interactions. Curr Opin Microbiol 53, 71–77 (2020). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources