

NGS-Based Genetic Analysis in a Cohort of Italian Patients with Suspected Inherited Myopathies and/or HyperCKemia
- PMID: 37510298
- PMCID: PMC10379733
- DOI: 10.3390/genes14071393
NGS-Based Genetic Analysis in a Cohort of Italian Patients with Suspected Inherited Myopathies and/or HyperCKemia
Abstract
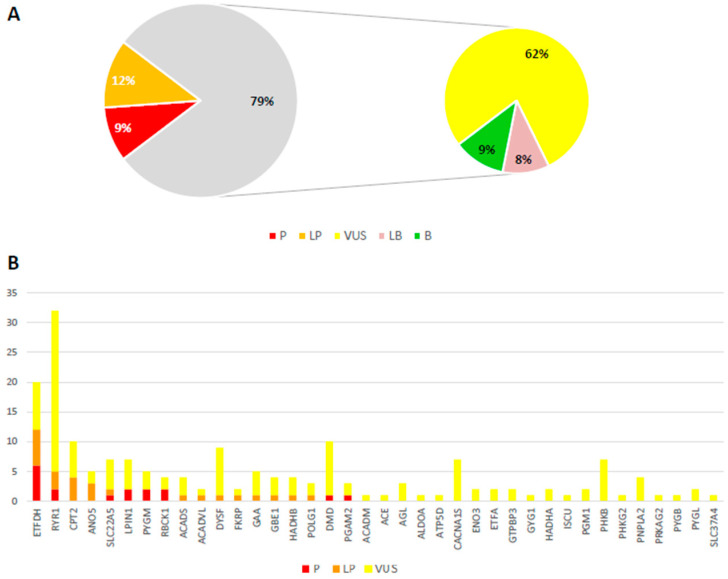
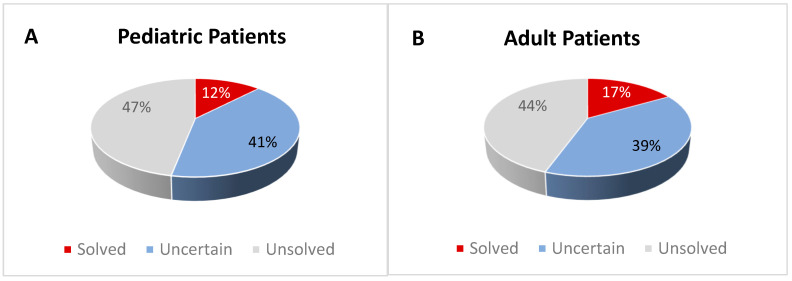
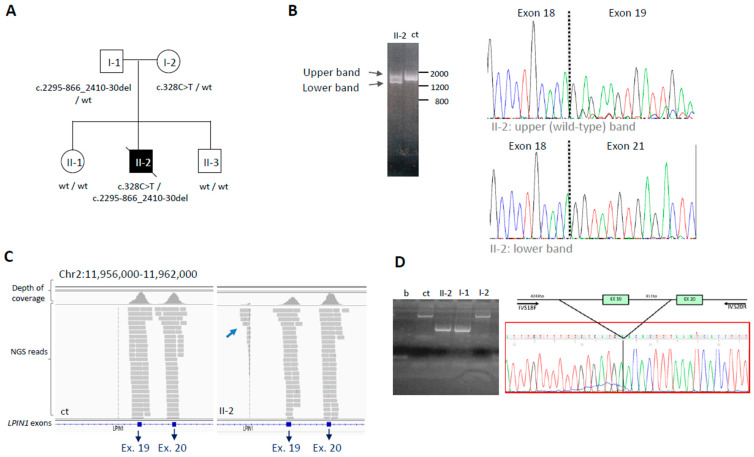
Introduction/Aims HyperCKemia is considered a hallmark of neuromuscular diseases. It can be either isolated or associated with cramps, myalgia, weakness, myoglobinuria, or rhabdomyolysis, suggesting a metabolic myopathy. The aim of this work was to investigate possible genetic causes in order to help diagnose patients with recurrent hyperCKemia or clinical suspicion of inherited metabolic myopathy. Methods A cohort of 139 patients (90 adults and 49 children) was analyzed using a custom panel containing 54 genes associated with hyperCKemia. Results A definite genetic diagnosis was obtained in 15.1% of cases, while candidate variants or variants of uncertain significance were found in a further 39.5%. Similar percentages were obtained in patients with infantile or adult onset, with some different causative genes. RYR1 was the gene most frequently identified, either with single or compound heterozygous variants, while ETFDH variants were the most common cause for recessive cases. In one patient, mRNA analysis allowed identifying a large LPIN1 deletion missed by DNA sequencing, leading to a certain diagnosis. Conclusion These data confirm the high genetic heterogeneity of hyperCKemia and metabolic myopathies. The reduced diagnostic yield suggests the existence of additional genes associated with this condition but also allows speculation that a significant number of cases presenting with hyperCKemia or muscle symptoms are due to extrinsic, not genetic, factors.
Keywords: Next Generation Sequencing (NGS); creatine kinase; hyperCKemia; myoglobinuria; rhabdomyolysis; skeletal muscle damage.
Conflict of interest statement
The authors declare no conflicst of interest.
Figures
References
-
- Prelle A., Tancredi L., Sciacco M., Chiveri L., Comi G.P., Martinelli-Boneschi F.M., Bagnardi V., Battistel A., Ciscato P., Bordoni A., et al. Retrospective study of a large population of patients with asymptomatic or minimally symptomatic raised serum creatine kinase levels. J. Neurol. 2002;249:305–311. doi: 10.1007/s004150200010. - DOI - PubMed
-
- Rowland L.P., Willner J., DiMauro S., Miranda A. Approaches to the membrane theory of Duchenne muscular dystrophy. In: Angelini C., Danieli G.A., Fontanari D., editors. Muscular Dystrophy-Advances and New Trends. Excerpta Medica; Amsterdam, The Netherlands: 1980. pp. 3–13.
-
- Khan F.Y. Rhabdomyolysis: A review of the literature. Neth. J. Med. 2009;67:272–283. - PubMed
