

Thrombin-Induced Microglia Activation Modulated through Aryl Hydrocarbon Receptors
- PMID: 37511175
- PMCID: PMC10380349
- DOI: 10.3390/ijms241411416
Thrombin-Induced Microglia Activation Modulated through Aryl Hydrocarbon Receptors
Abstract
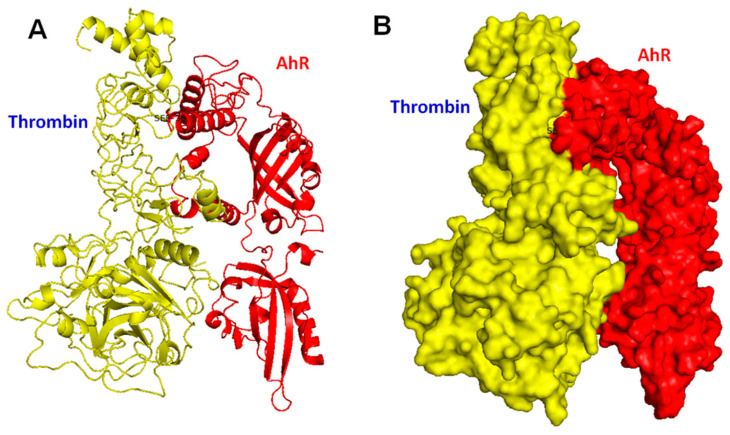
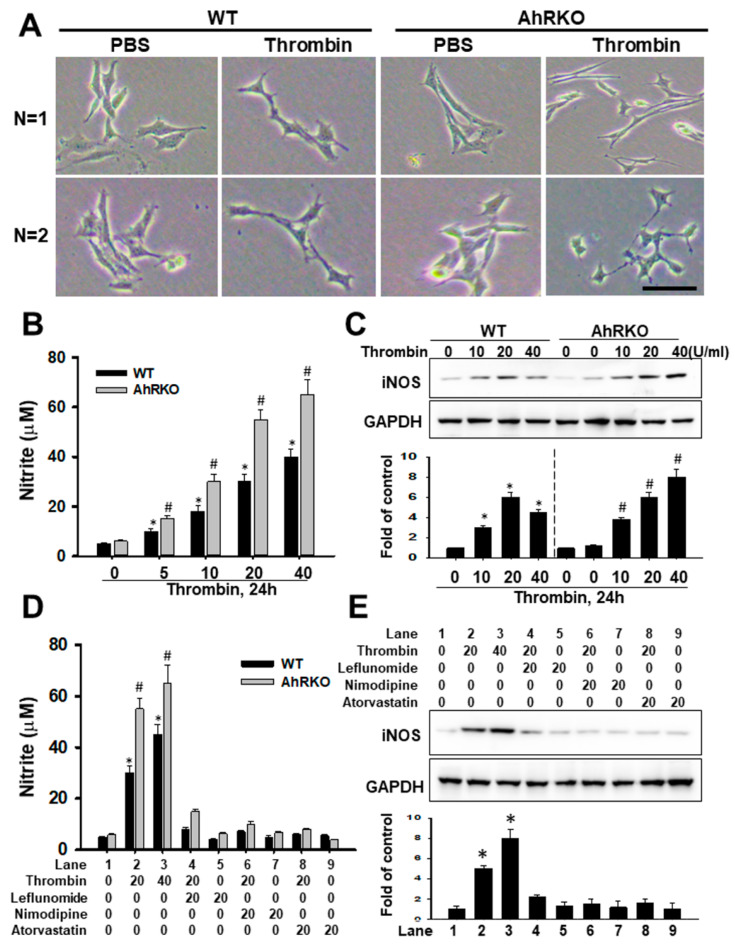
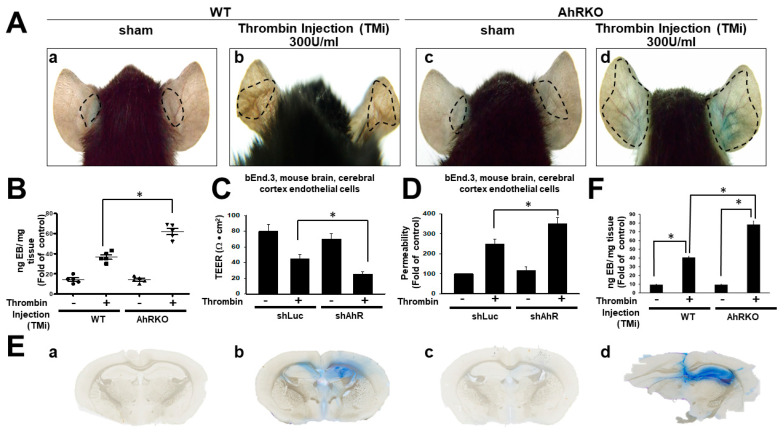
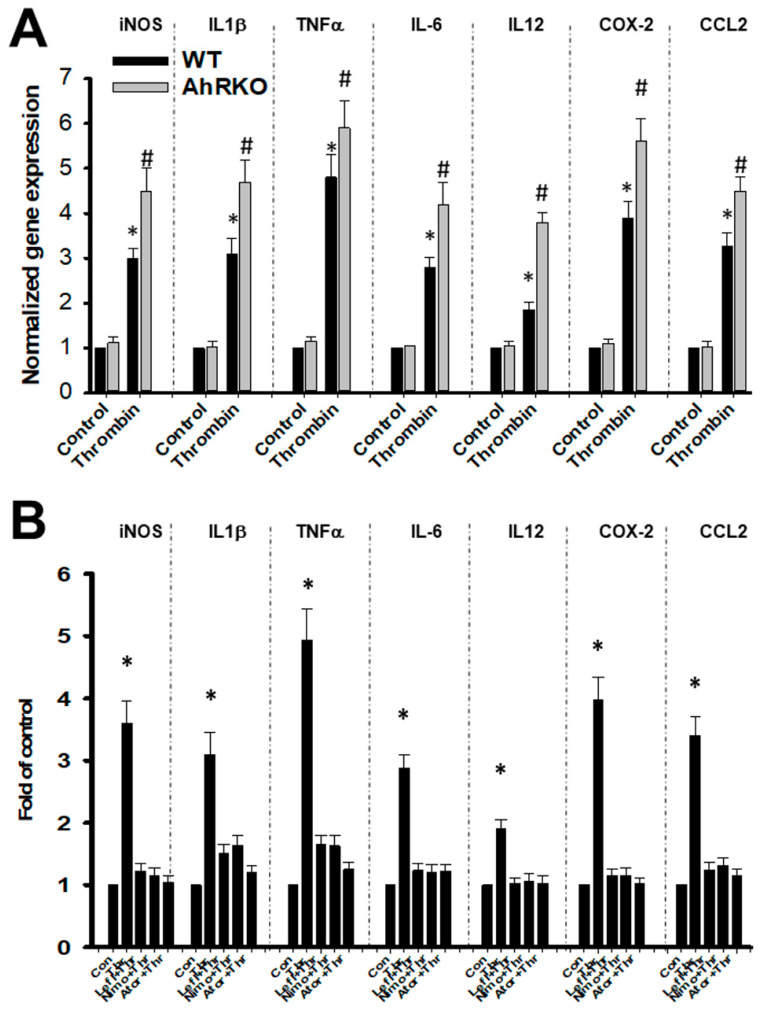
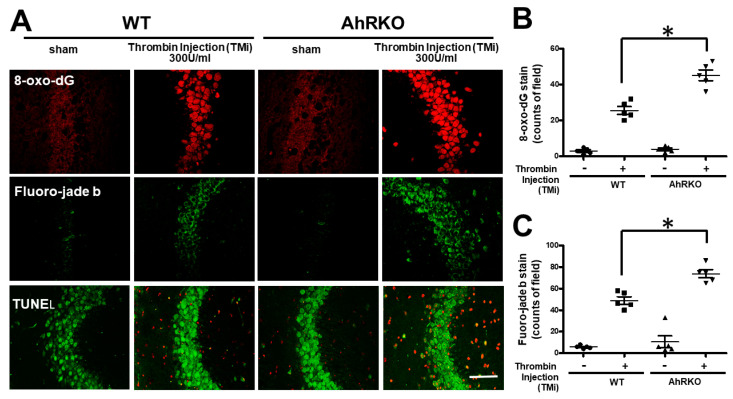
Thrombin is a multifunctional serine protein which is closely related to neurodegenerative disorders. The Aryl hydrocarbon receptor (AhR) is well expressed in microglia cells involving inflammatory disorders of the brain. However, it remains unclear as to how modulation of AhR expression by thrombin is related to the development of neurodegeneration disorders. In this study, we investigated the role of AhR in the development of thrombin-induced neurodegenerative processes, especially those concerning microglia. The primary culture of either wild type or AhR deleted microglia, as well as BV-2 cell lines, was used for an in vitro study. Hippocampal slice culture and animals with either wild type or with AhR deleted were used for the ex vivo and in vivo studies. Simulations of ligand protein docking showed a strong integration between the thrombin and AhR. In thrombin-triggered microglia cells, deleting AhR escalated both the NO release and iNOS expression. Such effects were abolished by the administration of the AhR agonist. In thrombin-activated microglia cells, downregulating AhR increased the following: vascular permeability, pro-inflammatory genetic expression, MMP-9 activity, and the ratio of M1/M2 phenotype. In the in vivo study, thrombin induced the activation of microglia and their volume, thereby contributing to the deterioration of neurobehavior. Deleting AhR furthermore aggravated the response in terms of impaired neurobehavior, increasing brain edema, aggregating microglia, and increasing neuronal death. In conclusion, thrombin caused the activation of microglia through increased vessel permeability, expression of inflammatory response, and phenotype of M1 microglia, as well the MMP activity. Deleting AhR augmented the above detrimental effects. These findings indicate that the modulation of AhR is essential for the regulation of thrombin-induced brain damages and that the AhR agonist may harbor the potentially therapeutic effect in thrombin-induced neurodegenerative disorder.
Keywords: aryl hydrocarbon receptor; inflammation; microglia; neurodegenerative disorder; thrombin.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
