

Inhibitor Trapping in N-Myristoyltransferases as a Mechanism for Drug Potency
- PMID: 37511367
- PMCID: PMC10380619
- DOI: 10.3390/ijms241411610
Inhibitor Trapping in N-Myristoyltransferases as a Mechanism for Drug Potency
Abstract
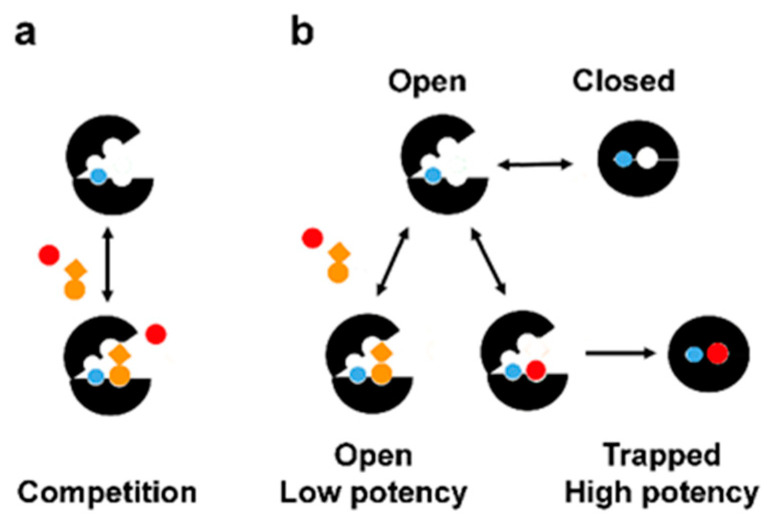
Predicting inhibitor potency is critical in drug design and development, yet it has remained one of computational biology's biggest unresolved challenges. Here, we show that in the case of the N-myristoyltransferase (NMT), this problem could be traced to the mechanisms by which the NMT enzyme is inhibited. NMT adopts open or closed conformations necessary for orchestrating the different steps of the catalytic process. The results indicate that the potency of the NMT inhibitors is determined by their ability to stabilize the enzyme conformation in the closed state, and that in this state, the small molecules themselves are trapped and locked inside the structure of the enzyme, creating a significant barrier for their dissociation. By using molecular dynamics simulations, we demonstrate that the conformational stabilization of the protein molecule in its closed form is highly correlated with the ligands activity and can be used to predict their potency. Hence, predicting inhibitor potency in silico might depend on modeling the conformational changes of the protein molecule upon binding of the ligand rather than estimating the changes in free binding energy that arise from their interaction.
Keywords: N-myristoyltransferases; NMT; conformational stability and dynamics; drug design; drug potency; enzyme inhibition; inhibitor potency; inhibitor trap; mechanism of inhibition; predicting inhibitor activity.
Conflict of interest statement
The authors declare no conflict of interest. The funders had no role in the design of the study, in the collection, analysis, or interpretation of data, in the writing of the manuscript, or in the decision to publish the results.
Figures
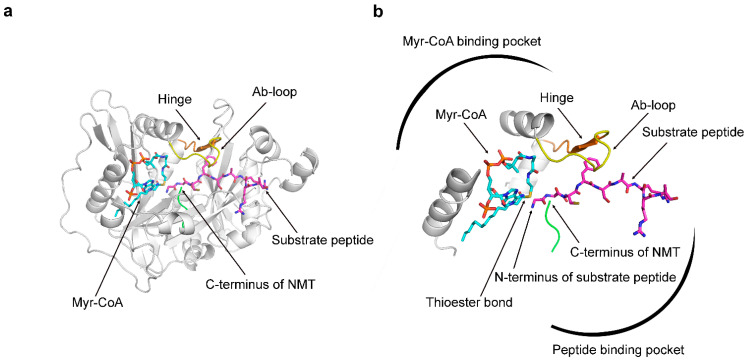
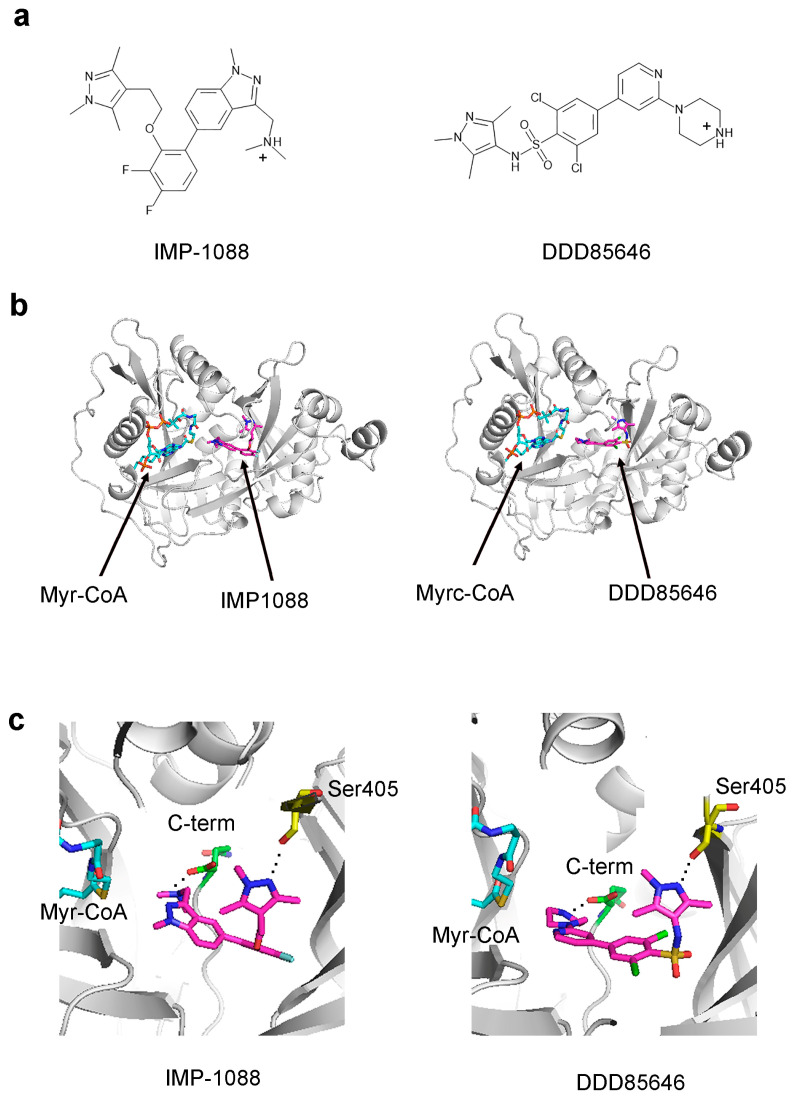
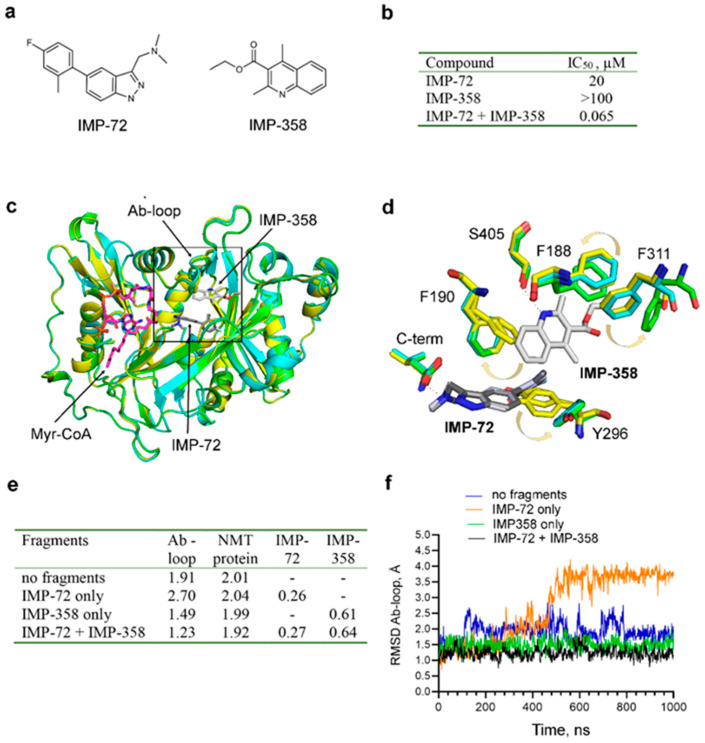
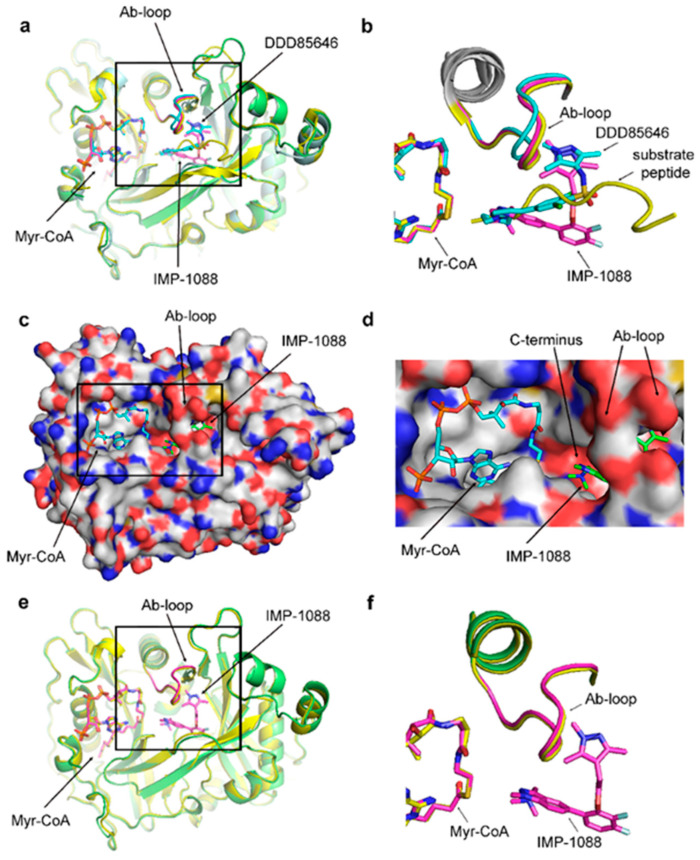
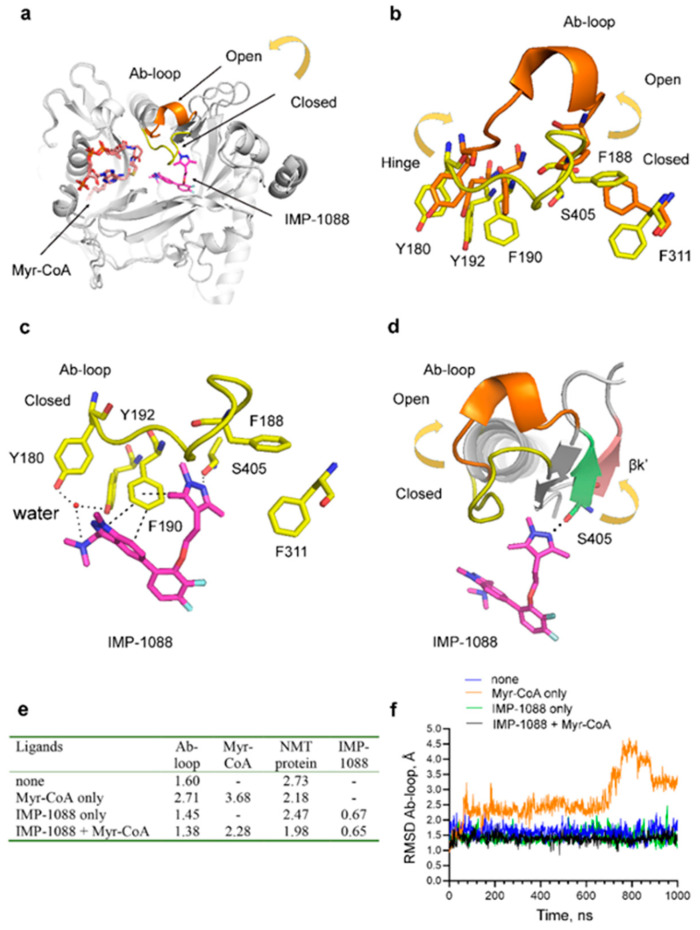
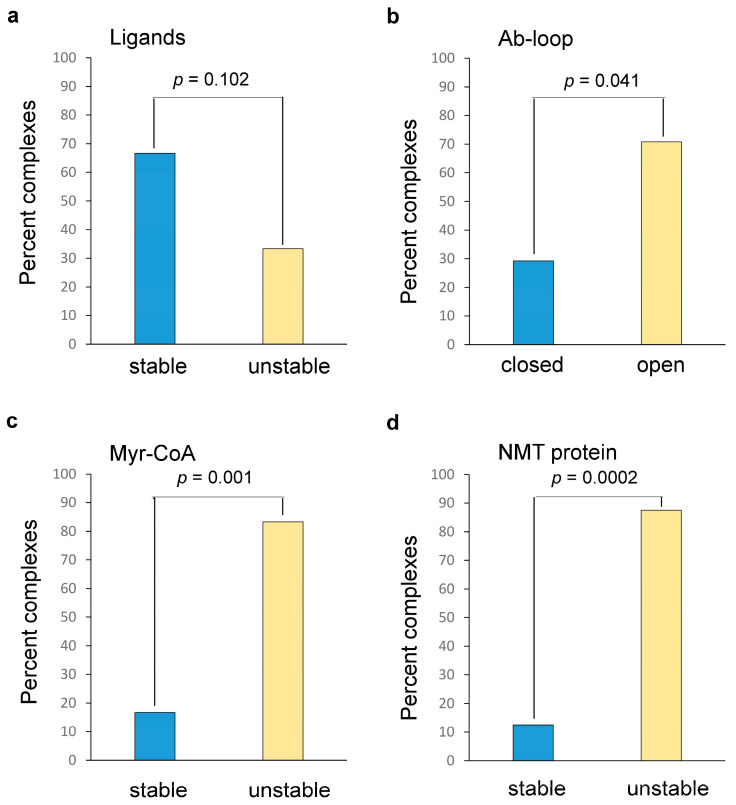
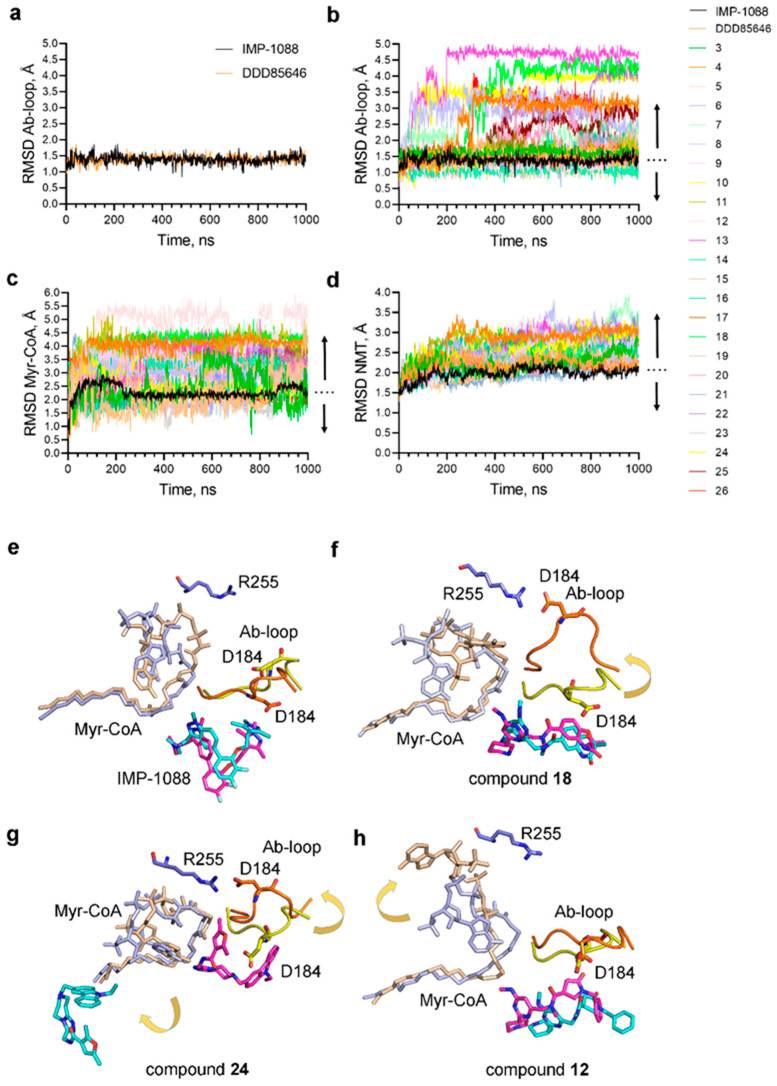
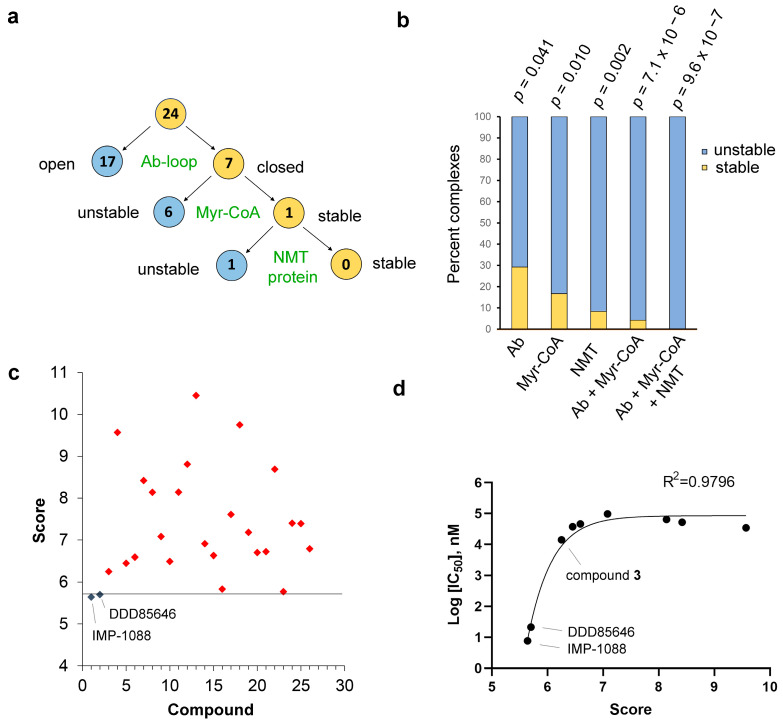
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
