

Neuromuscular disease genetics in under-represented populations: increasing data diversity
- PMID: 37516995
- PMCID: PMC10690022
- DOI: 10.1093/brain/awad254
Neuromuscular disease genetics in under-represented populations: increasing data diversity
Abstract
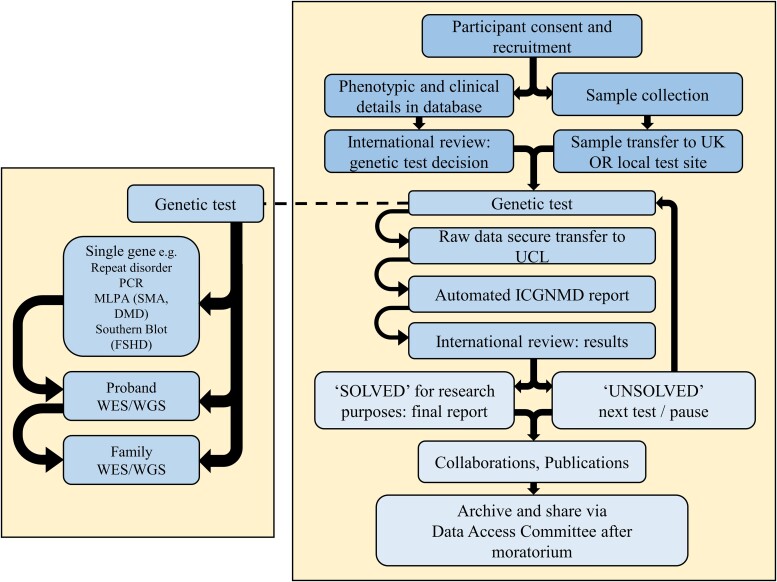
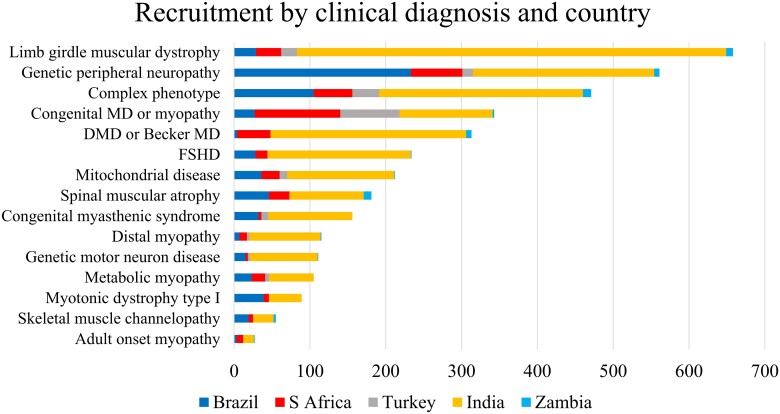
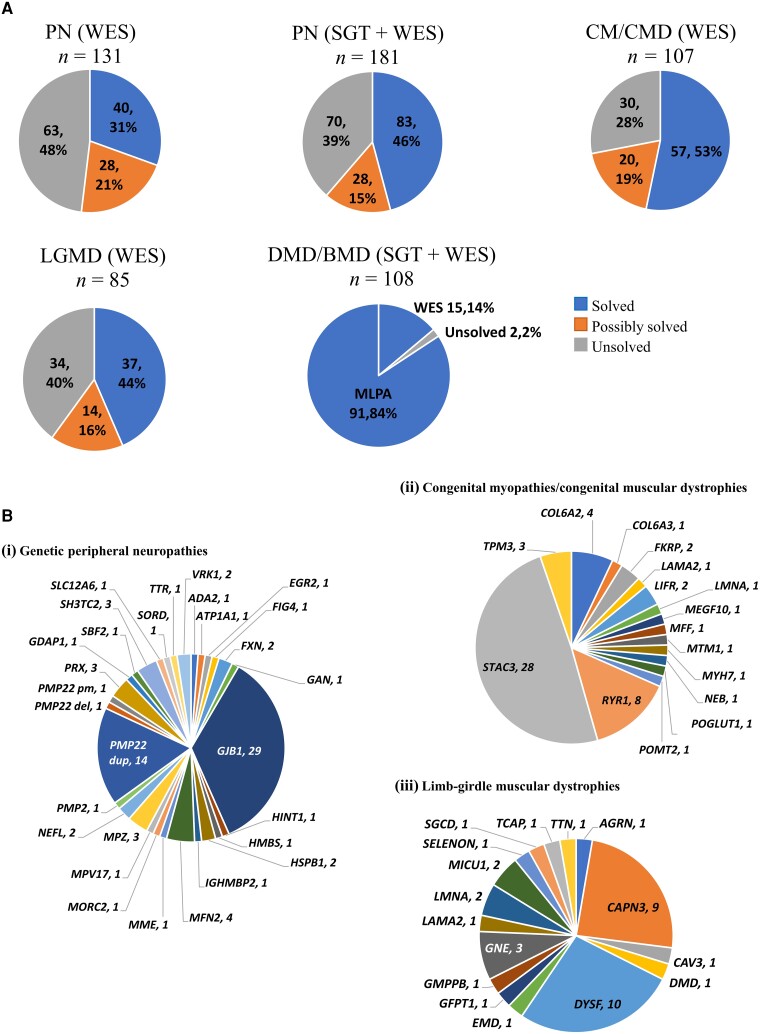
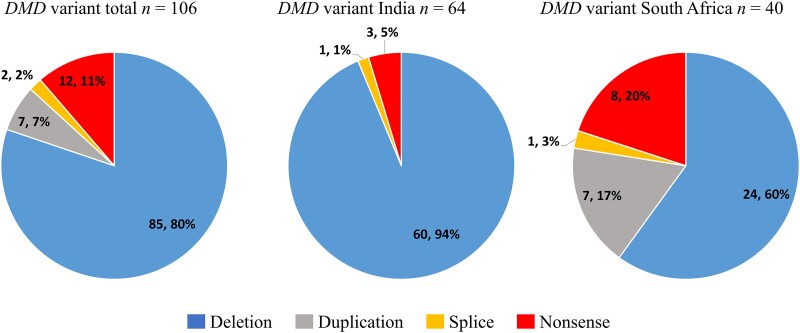
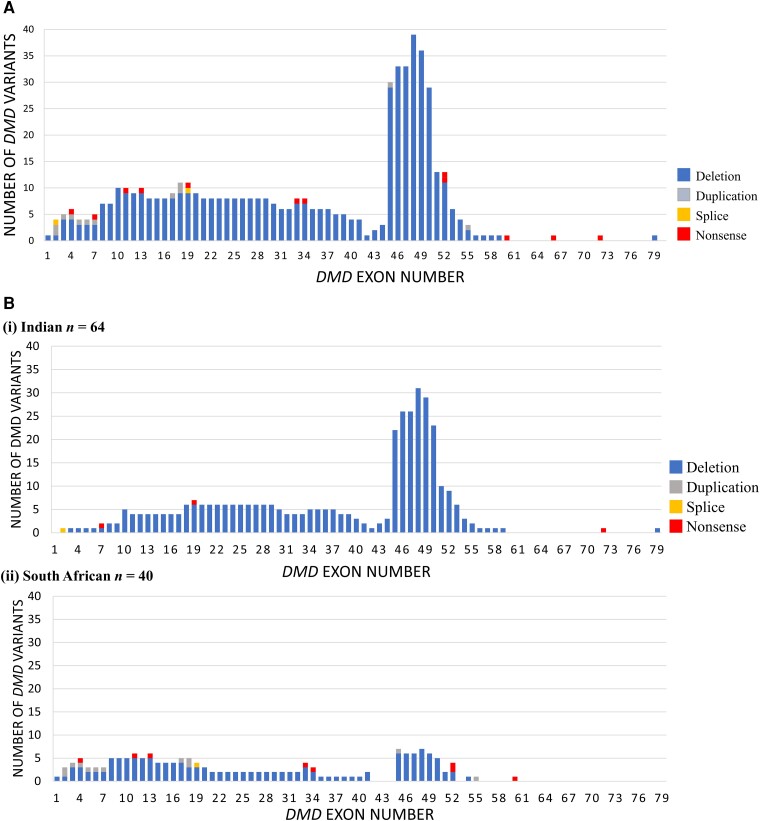
Neuromuscular diseases (NMDs) affect ∼15 million people globally. In high income settings DNA-based diagnosis has transformed care pathways and led to gene-specific therapies. However, most affected families are in low-to-middle income countries (LMICs) with limited access to DNA-based diagnosis. Most (86%) published genetic data is derived from European ancestry. This marked genetic data inequality hampers understanding of genetic diversity and hinders accurate genetic diagnosis in all income settings. We developed a cloud-based transcontinental partnership to build diverse, deeply-phenotyped and genetically characterized cohorts to improve genetic architecture knowledge, and potentially advance diagnosis and clinical management. We connected 18 centres in Brazil, India, South Africa, Turkey, Zambia, Netherlands and the UK. We co-developed a cloud-based data solution and trained 17 international neurology fellows in clinical genomic data interpretation. Single gene and whole exome data were analysed via a bespoke bioinformatics pipeline and reviewed alongside clinical and phenotypic data in global webinars to inform genetic outcome decisions. We recruited 6001 participants in the first 43 months. Initial genetic analyses 'solved' or 'possibly solved' ∼56% probands overall. In-depth genetic data review of the four commonest clinical categories (limb girdle muscular dystrophy, inherited peripheral neuropathies, congenital myopathy/muscular dystrophies and Duchenne/Becker muscular dystrophy) delivered a ∼59% 'solved' and ∼13% 'possibly solved' outcome. Almost 29% of disease causing variants were novel, increasing diverse pathogenic variant knowledge. Unsolved participants represent a new discovery cohort. The dataset provides a large resource from under-represented populations for genetic and translational research. In conclusion, we established a remote transcontinental partnership to assess genetic architecture of NMDs across diverse populations. It supported DNA-based diagnosis, potentially enabling genetic counselling, care pathways and eligibility for gene-specific trials. Similar virtual partnerships could be adopted by other areas of global genomic neurological practice to reduce genetic data inequality and benefit patients globally.
Keywords: capacity building; equality and diversity; genomic medicine; inherited neuromuscular disease; low-to-middle income country.
© The Author(s) 2023. Published by Oxford University Press on behalf of the Guarantors of Brain.
Conflict of interest statement
The authors report no competing interests.
Figures
References
-
- Deenen JC, Horlings CG, Verschuuren JJ, Verbeek AL, van Engelen BG. The epidemiology of neuromuscular disorders: a comprehensive overview of the literature. J Neuromuscul Dis. 2015;2:73–85. - PubMed
-
- Mercuri E, Pera MC, Scoto M, Finkel R, Muntoni F. Spinal muscular atrophy—insights and challenges in the treatment era. Nat Rev Neurol. 2020;16:706–715. - PubMed
-
- Verhaart IEC, Aartsma-Rus A. Therapeutic developments for Duchenne muscular dystrophy. Nat Rev Neurol. 2019;15:373–386. - PubMed
-
- Pereira L, Mutesa L, Tindana P, Ramsay M. African genetic diversity and adaptation inform a precision medicine agenda. Nat Rev Genet. 2021;22:284–306. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
