

Patterns of transcription factor binding and epigenome at promoters allow interpretable predictability of multiple functions of non-coding and coding genes
- PMID: 37520281
- PMCID: PMC10371796
- DOI: 10.1016/j.csbj.2023.07.014
Patterns of transcription factor binding and epigenome at promoters allow interpretable predictability of multiple functions of non-coding and coding genes
Abstract
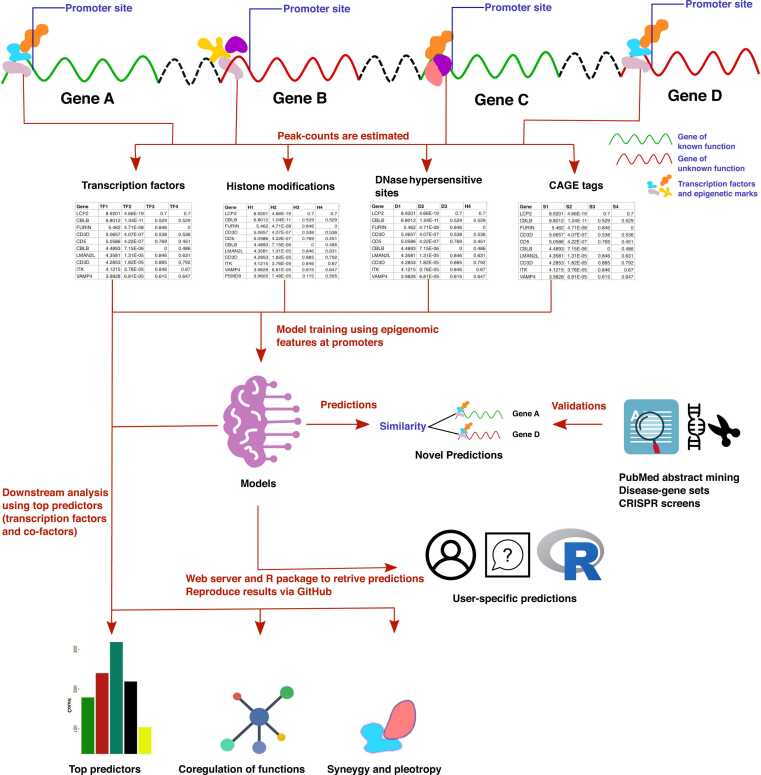
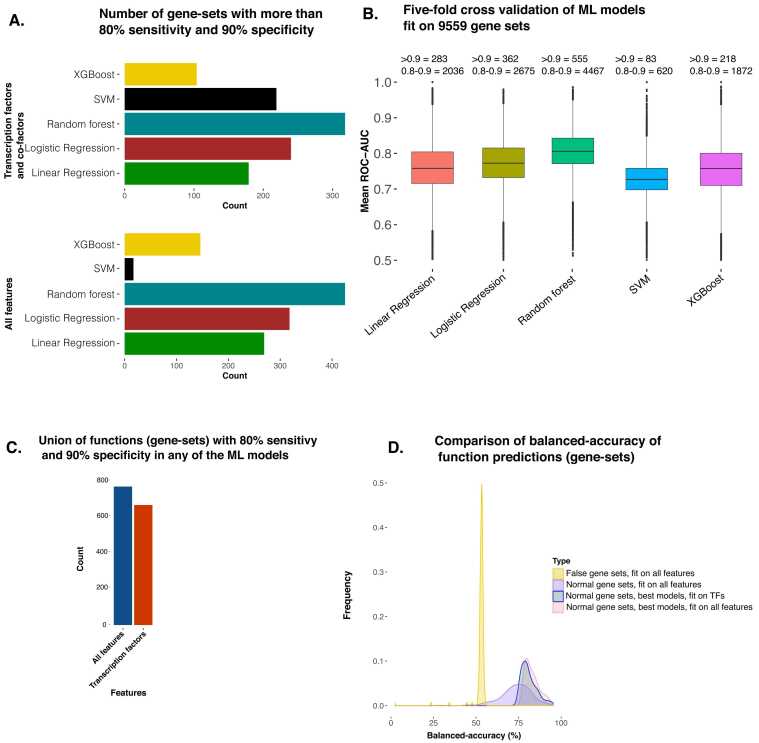
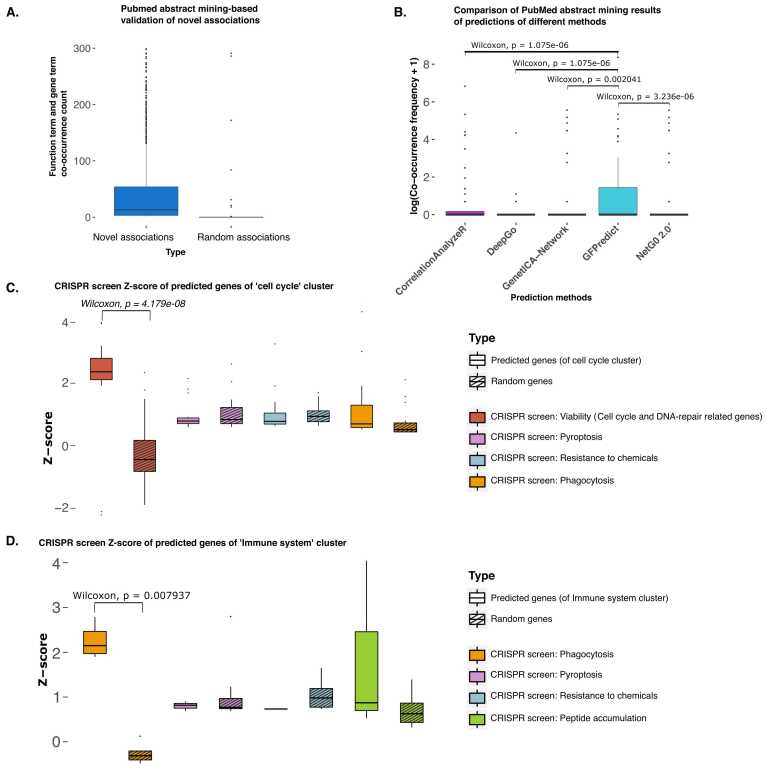
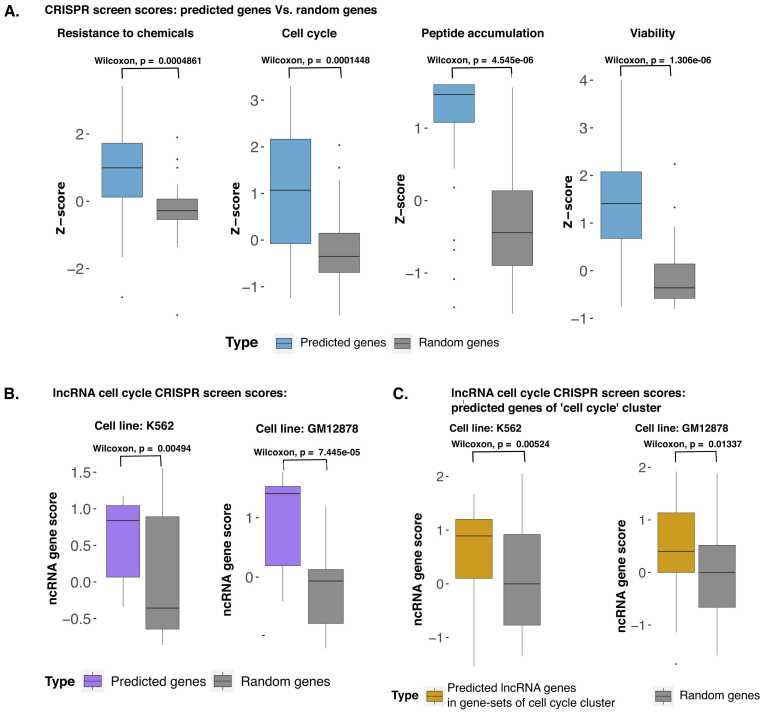
Understanding the biological roles of all genes only through experimental methods is challenging. A computational approach with reliable interpretability is needed to infer the function of genes, particularly for non-coding RNAs. We have analyzed genomic features that are present across both coding and non-coding genes like transcription factor (TF) and cofactor ChIP-seq (823), histone modifications ChIP-seq (n = 621), cap analysis gene expression (CAGE) tags (n = 255), and DNase hypersensitivity profiles (n = 255) to predict ontology-based functions of genes. Our approach for gene function prediction was reliable (>90% balanced accuracy) for 486 gene-sets. PubMed abstract mining and CRISPR screens supported the inferred association of genes with biological functions, for which our method had high accuracy. Further analysis revealed that TF-binding patterns at promoters have high predictive strength for multiple functions. TF-binding patterns at the promoter add an unexplored dimension of explainable regulatory aspects of genes and their functions. Therefore, we performed a comprehensive analysis for the functional-specificity of TF-binding patterns at promoters and used them for clustering functions to reveal many latent groups of gene-sets involved in common major cellular processes. We also showed how our approach could be used to infer the functions of non-coding genes using the CRISPR screens of coding genes, which were validated using a long non-coding RNA CRISPR screen. Thus our results demonstrated the generality of our approach by using gene-sets from CRISPR screens. Overall, our approach opens an avenue for predicting the involvement of non-coding genes in various functions.
Keywords: Coregulation of functions; Epigenetics; Functional genomics; Gene function prediction; Gene regulation; General transcription factor (GTF); LncRNA); Long noncoding RNA (long ncRNA.
© 2023 The Authors.
Conflict of interest statement
Author declare no conflict of interest.
Figures



References
LinkOut - more resources
Full Text Sources
Miscellaneous