

Integrative human atrial modelling unravels interactive protein kinase A and Ca2+/calmodulin-dependent protein kinase II signalling as key determinants of atrial arrhythmogenesis
- PMID: 37523735
- PMCID: PMC11318383
- DOI: 10.1093/cvr/cvad118
Integrative human atrial modelling unravels interactive protein kinase A and Ca2+/calmodulin-dependent protein kinase II signalling as key determinants of atrial arrhythmogenesis
Abstract
Aims: Atrial fibrillation (AF), the most prevalent clinical arrhythmia, is associated with atrial remodelling manifesting as acute and chronic alterations in expression, function, and regulation of atrial electrophysiological and Ca2+-handling processes. These AF-induced modifications crosstalk and propagate across spatial scales creating a complex pathophysiological network, which renders AF resistant to existing pharmacotherapies that predominantly target transmembrane ion channels. Developing innovative therapeutic strategies requires a systems approach to disentangle quantitatively the pro-arrhythmic contributions of individual AF-induced alterations.
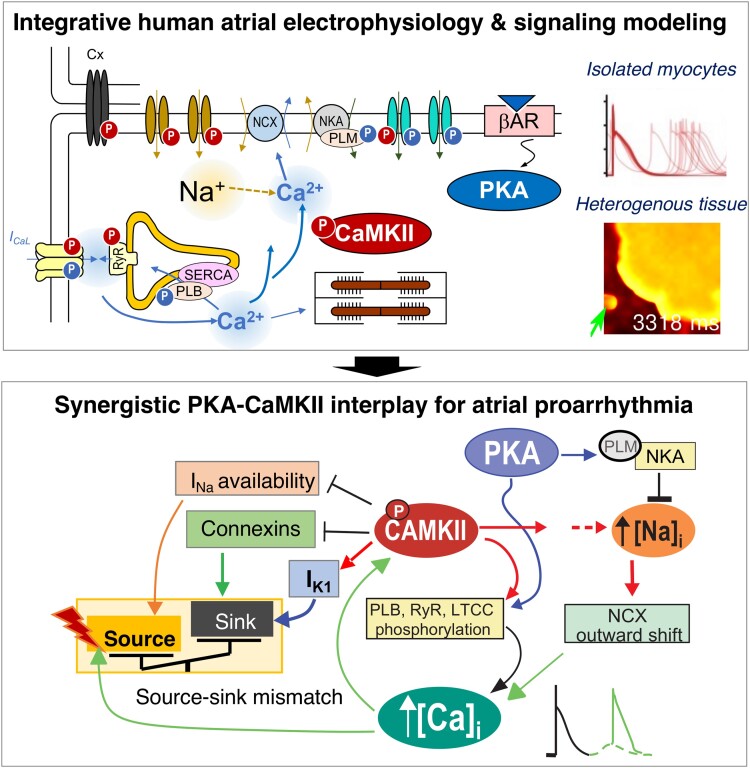
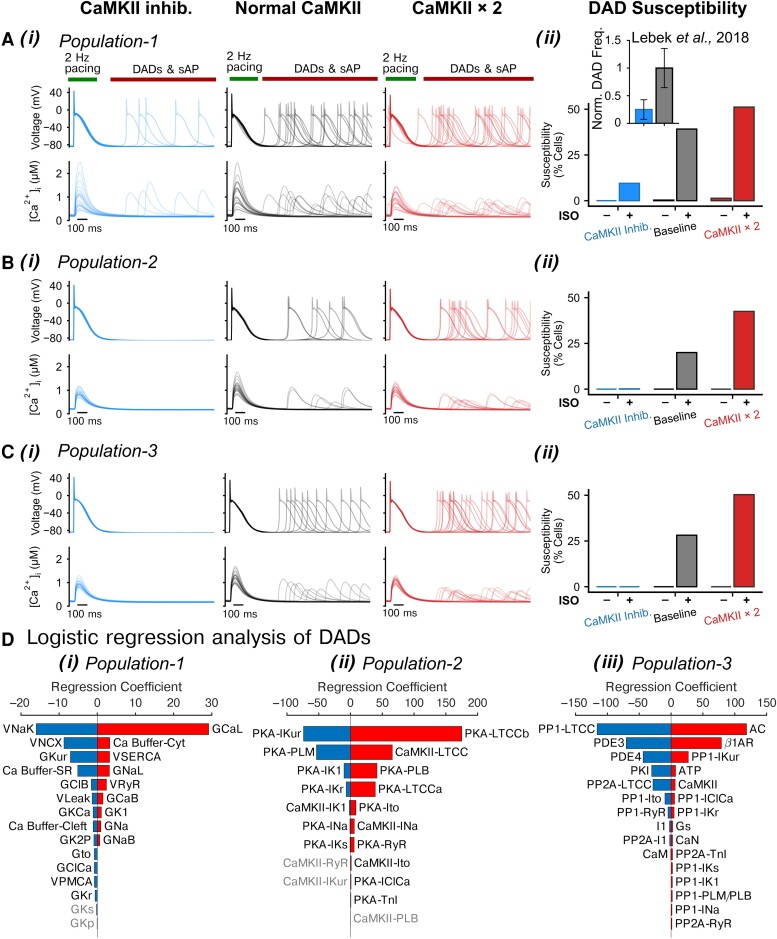
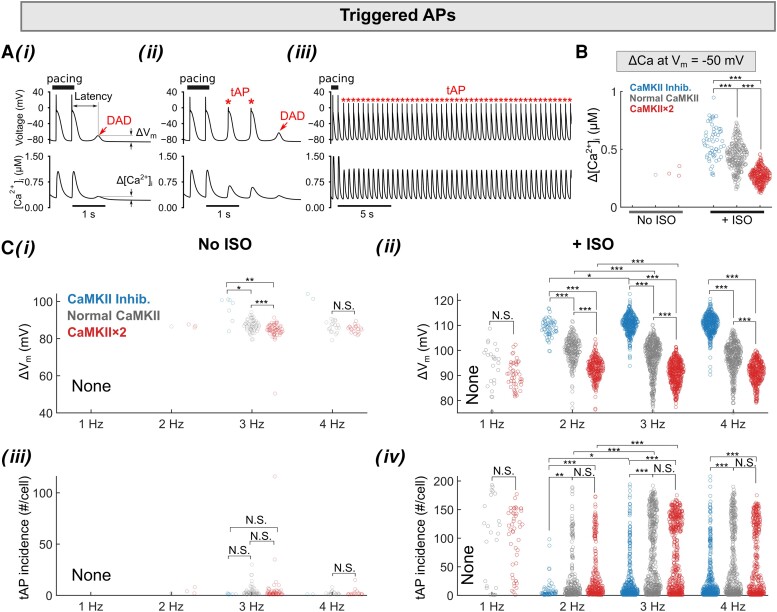
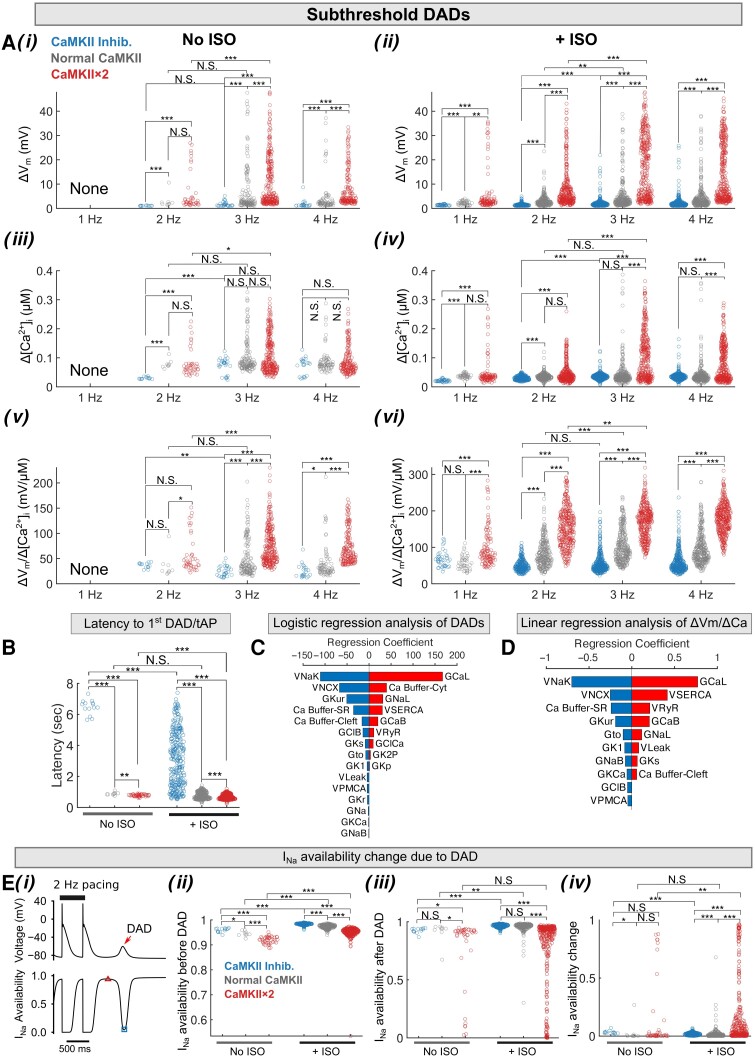
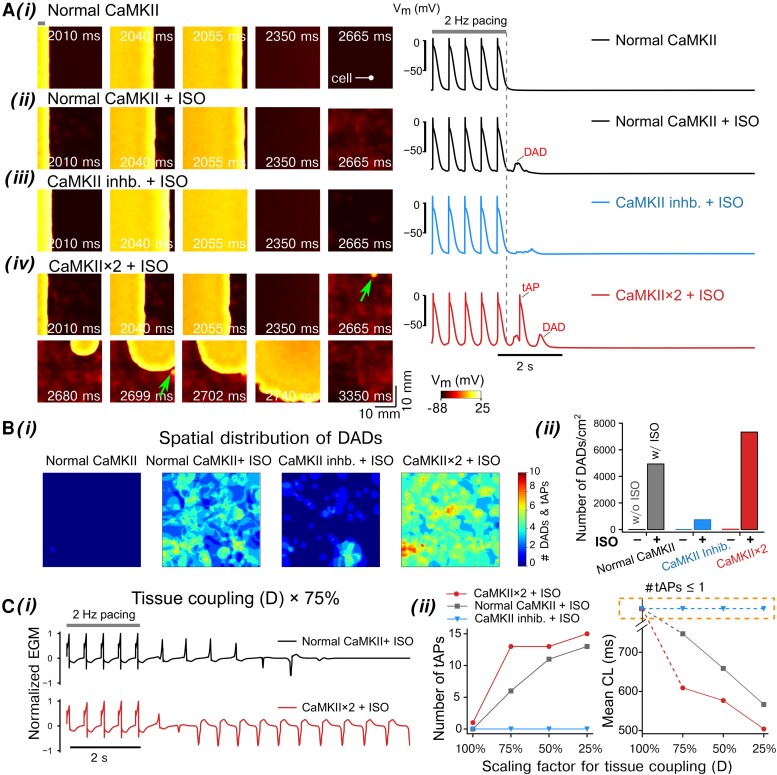
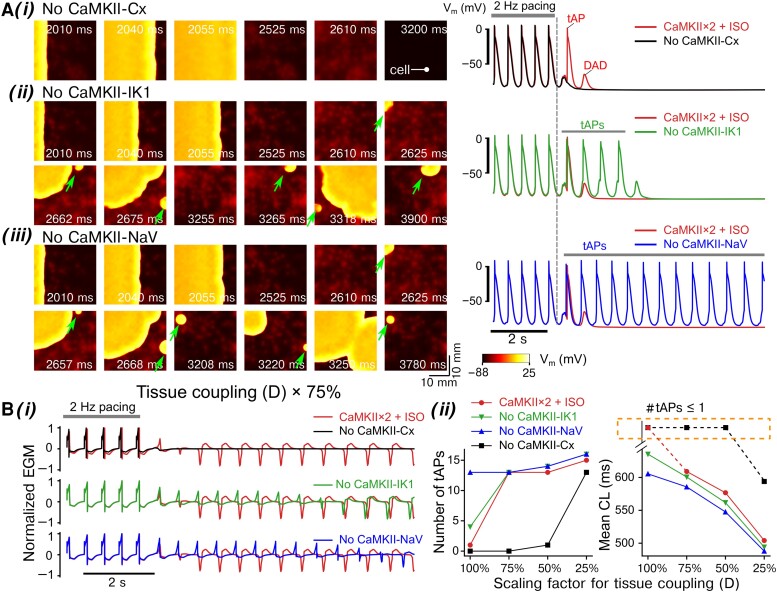
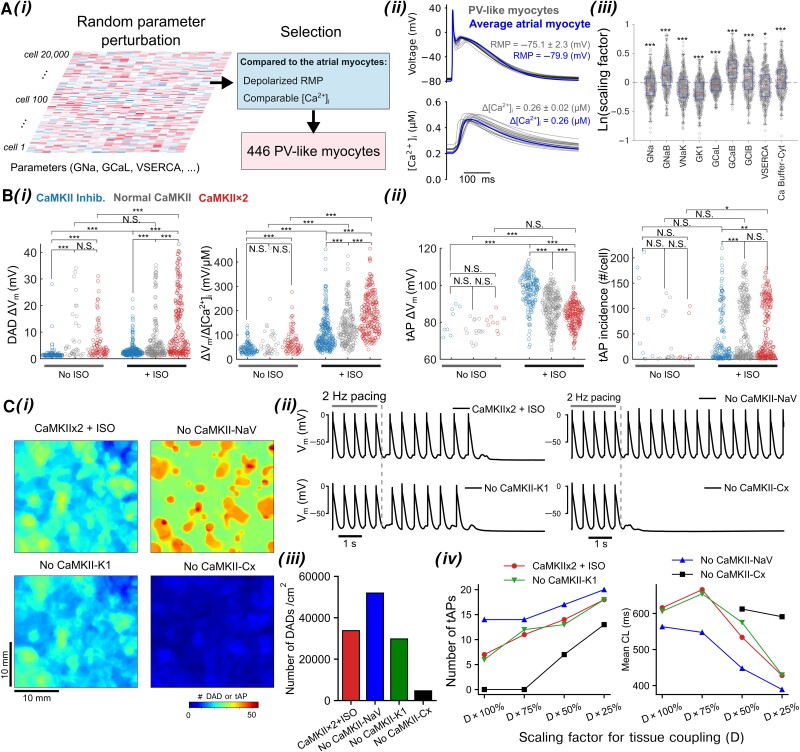
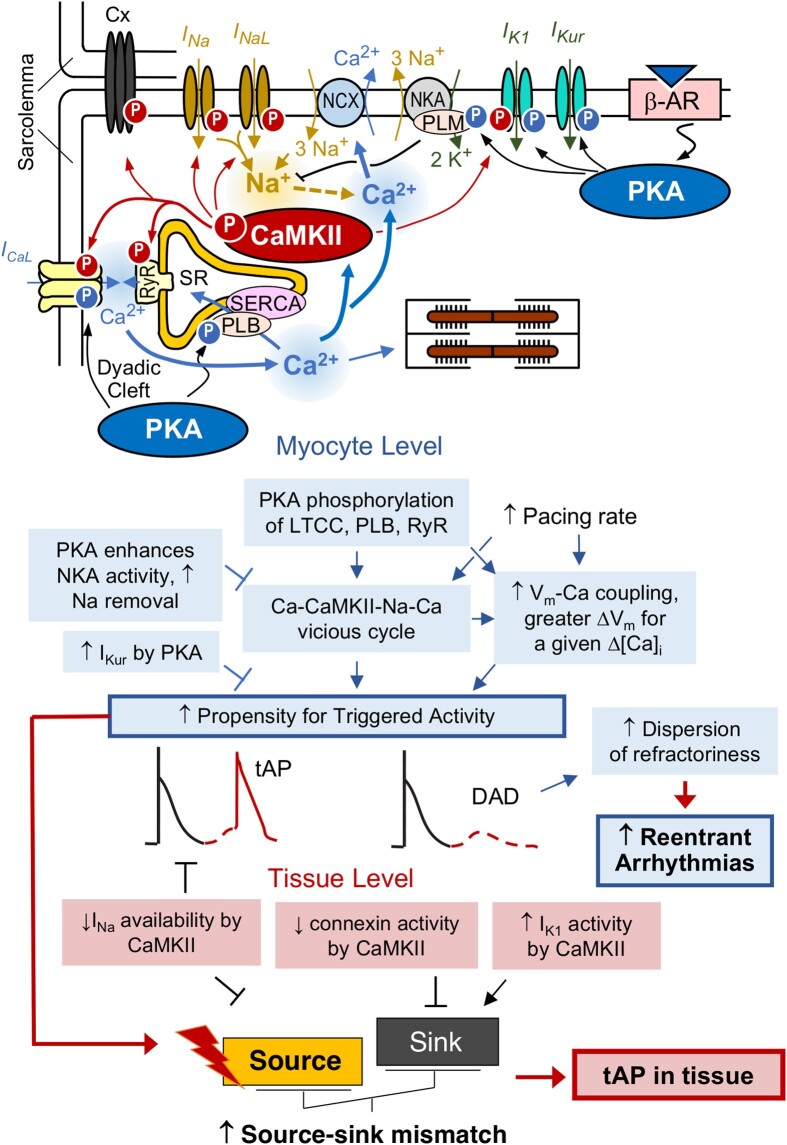
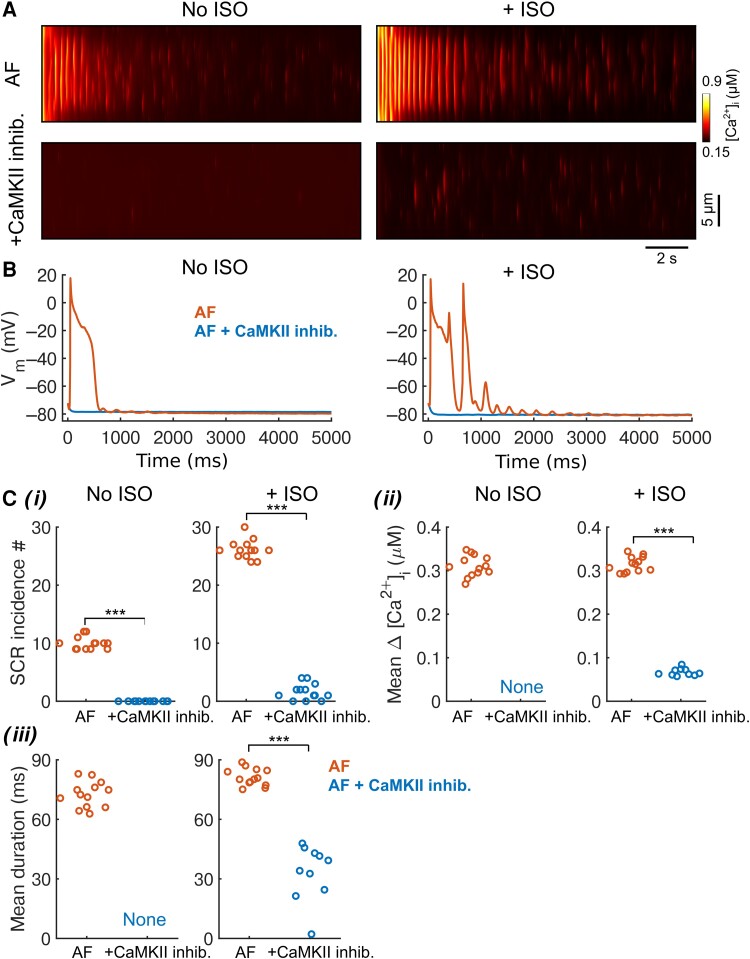
Methods and results: Here, we built a novel computational framework for simulating electrophysiology and Ca2+-handling in human atrial cardiomyocytes and tissues, and their regulation by key upstream signalling pathways [i.e. protein kinase A (PKA), and Ca2+/calmodulin-dependent protein kinase II (CaMKII)] involved in AF-pathogenesis. Populations of atrial cardiomyocyte models were constructed to determine the influence of subcellular ionic processes, signalling components, and regulatory networks on atrial arrhythmogenesis. Our results reveal a novel synergistic crosstalk between PKA and CaMKII that promotes atrial cardiomyocyte electrical instability and arrhythmogenic triggered activity. Simulations of heterogeneous tissue demonstrate that this cellular triggered activity is further amplified by CaMKII- and PKA-dependent alterations of tissue properties, further exacerbating atrial arrhythmogenesis.
Conclusions: Our analysis reveals potential mechanisms by which the stress-associated adaptive changes turn into maladaptive pro-arrhythmic triggers at the cellular and tissue levels and identifies potential anti-AF targets. Collectively, our integrative approach is powerful and instrumental to assemble and reconcile existing knowledge into a systems network for identifying novel anti-AF targets and innovative approaches moving beyond the traditional ion channel-based strategy.
Keywords: Arrhythmias; Atrial fibrillation; Computational biology; Electrophysiology; Physiology; Population modelling; Systems biology; Upstream signalling.
© The Author(s) 2023. Published by Oxford University Press on behalf of the European Society of Cardiology.
Conflict of interest statement
Conflict of interest: None declared.
Figures
Similar articles
-
Computational modelling of cardiac fibroblast signalling reveals a key role for Ca2+ in driving atrial fibrillation-associated fibrosis.J Physiol. 2025 Jun 19:10.1113/JP289040. doi: 10.1113/JP289040. Online ahead of print. J Physiol. 2025. PMID: 40538115
-
Interleukin-33/ST2 axis involvement in atrial remodeling and arrhythmogenesis.Transl Res. 2024 Jun;268:1-12. doi: 10.1016/j.trsl.2024.01.006. Epub 2024 Jan 18. Transl Res. 2024. PMID: 38244770
-
LNK/SH2B3 loss of function increases susceptibility to murine and human atrial fibrillation.Cardiovasc Res. 2024 Jul 2;120(8):899-913. doi: 10.1093/cvr/cvae036. Cardiovasc Res. 2024. PMID: 38377486 Free PMC article.
-
Mechanisms of stretch-induced electro-anatomical remodeling and atrial arrhythmogenesis.J Mol Cell Cardiol. 2024 Aug;193:11-24. doi: 10.1016/j.yjmcc.2024.05.011. Epub 2024 May 24. J Mol Cell Cardiol. 2024. PMID: 38797242 Free PMC article. Review.
-
Intravenous magnesium sulphate and sotalol for prevention of atrial fibrillation after coronary artery bypass surgery: a systematic review and economic evaluation.Health Technol Assess. 2008 Jun;12(28):iii-iv, ix-95. doi: 10.3310/hta12280. Health Technol Assess. 2008. PMID: 18547499
Cited by
-
Macrophage-mediated IL-6 signaling drives ryanodine receptor-2 calcium leak in postoperative atrial fibrillation.J Clin Invest. 2025 Mar 6;135(9):e187711. doi: 10.1172/JCI187711. eCollection 2025 May 1. J Clin Invest. 2025. PMID: 40048254 Free PMC article.
-
Dual effects of the small-conductance Ca2+-activated K+ current on human atrial electrophysiology and Ca2+-driven arrhythmogenesis: an in silico study.Am J Physiol Heart Circ Physiol. 2023 Oct 1;325(4):H896-H908. doi: 10.1152/ajpheart.00362.2023. Epub 2023 Aug 25. Am J Physiol Heart Circ Physiol. 2023. PMID: 37624096 Free PMC article.
-
Enhanced Ca2+-Driven Arrhythmogenic Events in Female Patients With Atrial Fibrillation: Insights From Computational Modeling.JACC Clin Electrophysiol. 2024 Nov;10(11):2371-2391. doi: 10.1016/j.jacep.2024.07.020. Epub 2024 Sep 25. JACC Clin Electrophysiol. 2024. PMID: 39340505
-
Beta-Adrenergic Activation of the Inward Rectifier K+ Current Is Mediated by the CaMKII Pathway in Canine Ventricular Cardiomyocytes.Int J Mol Sci. 2024 Oct 29;25(21):11609. doi: 10.3390/ijms252111609. Int J Mol Sci. 2024. PMID: 39519160 Free PMC article.
References
-
- Andrade J, Khairy P, Dobrev D, Nattel S. The clinical profile and pathophysiology of atrial fibrillation relationships among clinical features, epidemiology, and mechanisms. Circ Res 2014;114:1453–1468. - PubMed
-
- Chung MK, Refaat M, Shen W-K, Kutyifa V, Cha Y-M, Di Biase L, Baranchuk A, Lampert R, Natale A, Fisher J, Lakkireddy DR. Atrial fibrillation. J Am Coll Cardiol 2020;75:1689–1713. - PubMed
-
- Heijman J, Guichard J-B, Dobrev D, Nattel S. Translational challenges in atrial fibrillation. Circ Res 2018;122:752–773. - PubMed
-
- Michaud GF, Stevenson WG. Atrial fibrillation. N Engl J Med 2021;384:353–361. - PubMed
