

Bacterial-fungal interactions promote parallel evolution of global transcriptional regulators in a widespread Staphylococcus species
- PMID: 37524910
- PMCID: PMC10432416
- DOI: 10.1038/s41396-023-01462-5
Bacterial-fungal interactions promote parallel evolution of global transcriptional regulators in a widespread Staphylococcus species
Abstract
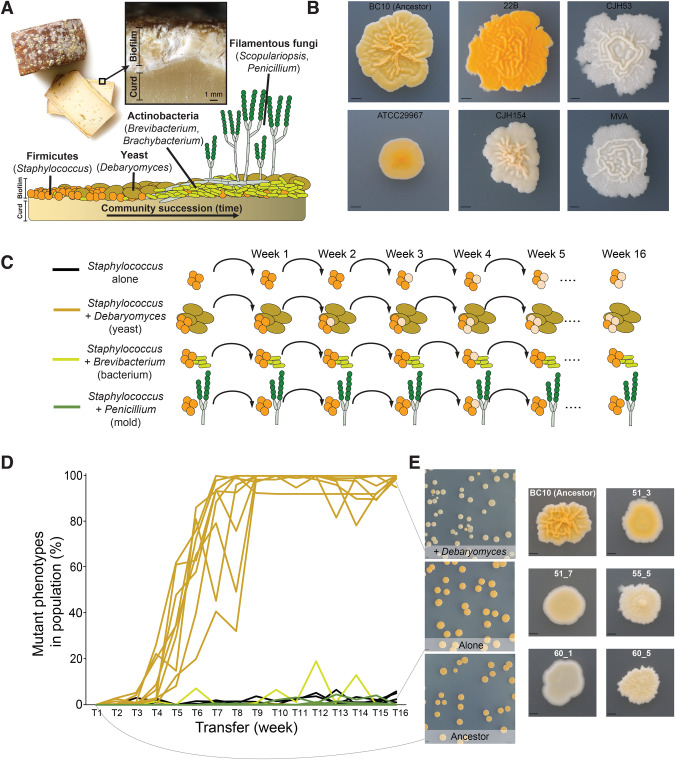
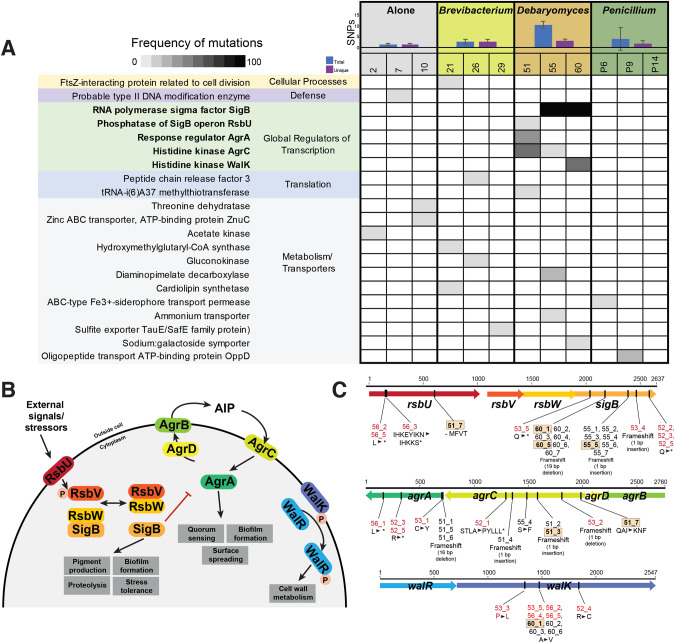
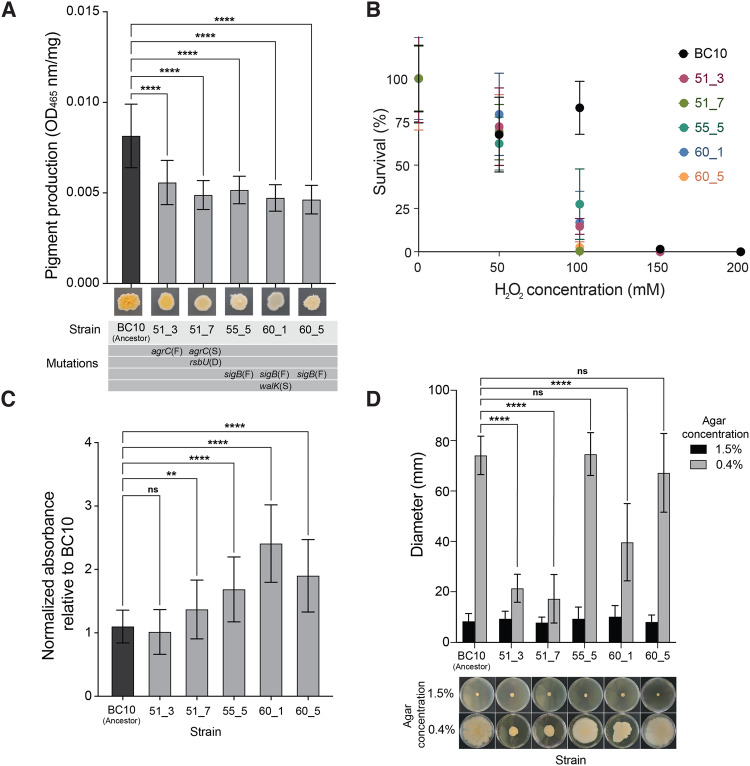
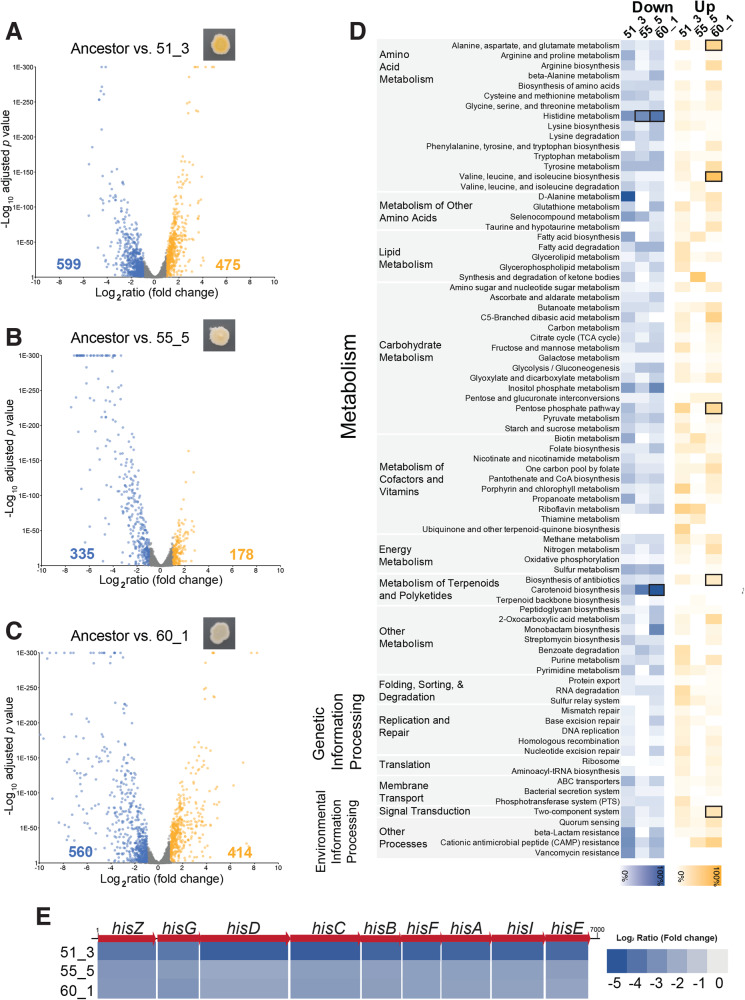
Experimental studies of microbial evolution have largely focused on monocultures of model organisms, but most microbes live in communities where interactions with other species may impact rates and modes of evolution. Using the cheese rind model microbial community, we determined how species interactions shape the evolution of the widespread food- and animal-associated bacterium Staphylococcus xylosus. We evolved S. xylosus for 450 generations alone or in co-culture with one of three microbes: the yeast Debaryomyces hansenii, the bacterium Brevibacterium aurantiacum, and the mold Penicillium solitum. We used the frequency of colony morphology mutants (pigment and colony texture phenotypes) and whole-genome sequencing of isolates to quantify phenotypic and genomic evolution. The yeast D. hansenii strongly promoted diversification of S. xylosus. By the end of the experiment, all populations co-cultured with the yeast were dominated by pigment and colony morphology mutant phenotypes. Populations of S. xylosus grown alone, with B. aurantiacum, or with P. solitum did not evolve novel phenotypic diversity. Whole-genome sequencing of individual mutant isolates across all four treatments identified numerous unique mutations in the operons for the SigB, Agr, and WalRK global regulators, but only in the D. hansenii treatment. Phenotyping and RNA-seq experiments highlighted altered pigment and biofilm production, spreading, stress tolerance, and metabolism of S. xylosus mutants. Fitness experiments revealed antagonistic pleiotropy, where beneficial mutations that evolved in the presence of the yeast had strong negative fitness effects in other biotic environments. This work demonstrates that bacterial-fungal interactions can have long-term evolutionary consequences within multispecies microbiomes by facilitating the evolution of strain diversity.
© 2023. The Author(s), under exclusive licence to International Society for Microbial Ecology.
Conflict of interest statement
The authors declare no competing interests.
Figures
Comment in
-
Diversification during cross-kingdom microbial experimental evolution.ISME J. 2023 Sep;17(9):1355-1357. doi: 10.1038/s41396-023-01479-w. Epub 2023 Jul 31. ISME J. 2023. PMID: 37524911 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
