

Genetic impacts on DNA methylation help elucidate regulatory genomic processes
- PMID: 37525248
- PMCID: PMC10391992
- DOI: 10.1186/s13059-023-03011-x
Genetic impacts on DNA methylation help elucidate regulatory genomic processes
Abstract
Background: Pinpointing genetic impacts on DNA methylation can improve our understanding of pathways that underlie gene regulation and disease risk.
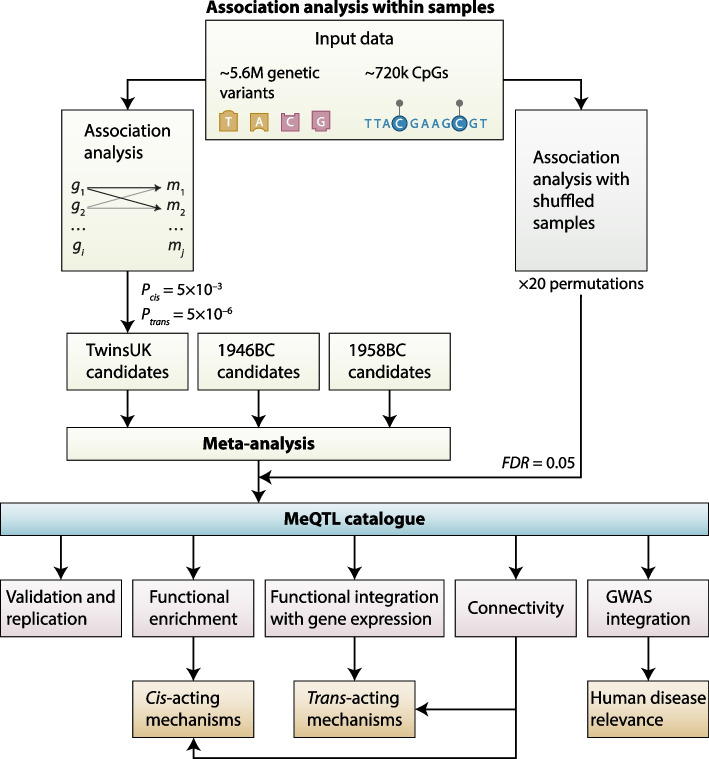
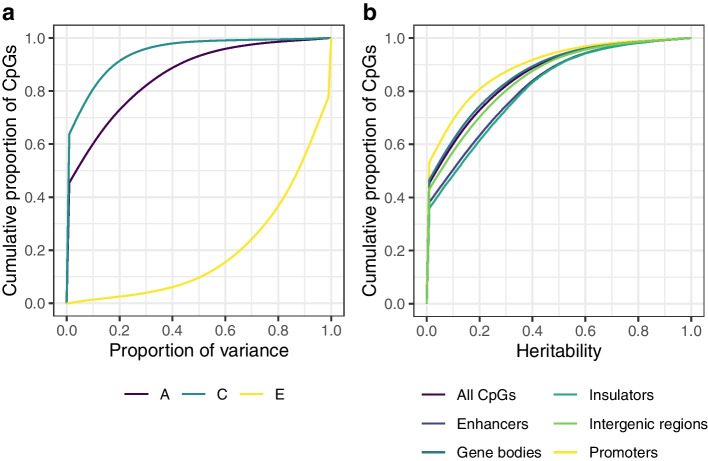
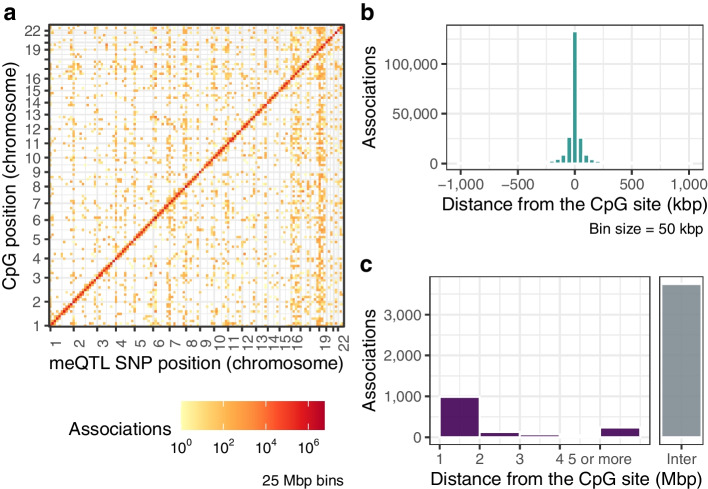
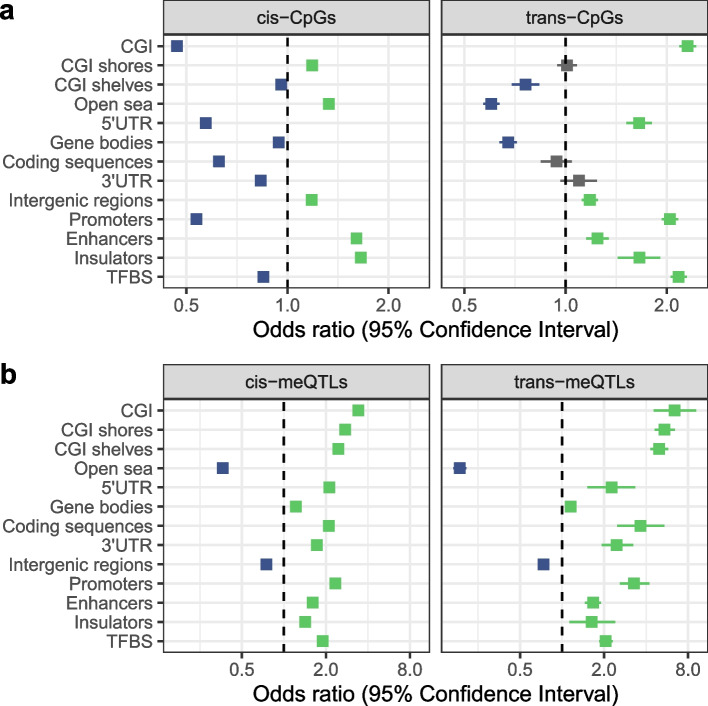
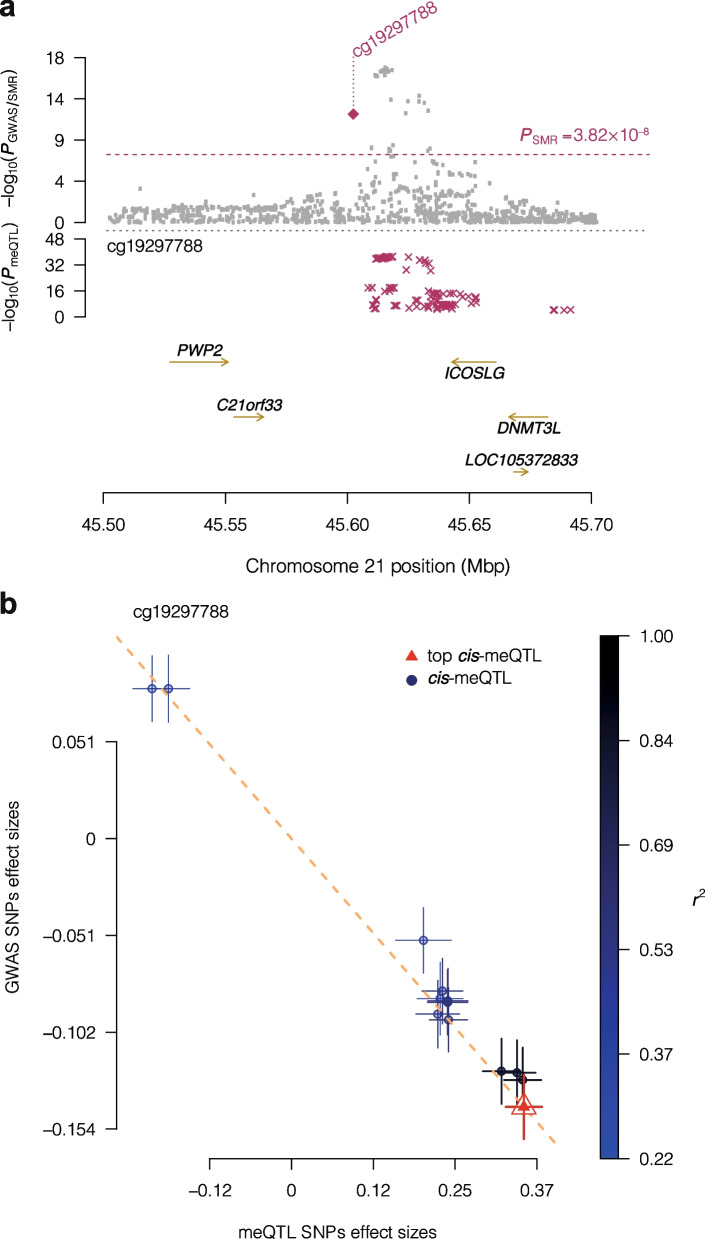
Results: We report heritability and methylation quantitative trait locus (meQTL) analysis at 724,499 CpGs profiled with the Illumina Infinium MethylationEPIC array in 2358 blood samples from three UK cohorts. Methylation levels at 34.2% of CpGs are affected by SNPs, and 98% of effects are cis-acting or within 1 Mbp of the tested CpG. Our results are consistent with meQTL analyses based on the former Illumina Infinium HumanMethylation450 array. Both SNPs and CpGs with meQTLs are overrepresented in enhancers, which have improved coverage on this platform compared to previous approaches. Co-localisation analyses across genetic effects on DNA methylation and 56 human traits identify 1520 co-localisations across 1325 unique CpGs and 34 phenotypes, including in disease-relevant genes, such as USP1 and DOCK7 (total cholesterol levels), and ICOSLG (inflammatory bowel disease). Enrichment analysis of meQTLs and integration with expression QTLs give insights into mechanisms underlying cis-meQTLs (e.g. through disruption of transcription factor binding sites for CTCF and SMC3) and trans-meQTLs (e.g. through regulating the expression of ACD and SENP7 which can modulate DNA methylation at distal sites).
Conclusions: Our findings improve the characterisation of the mechanisms underlying DNA methylation variability and are informative for prioritisation of GWAS variants for functional follow-ups. The MeQTL EPIC Database and viewer are available online at https://epicmeqtl.kcl.ac.uk .
Keywords: DNA methylation; GWAS; Heritability; Methylation quantitative trait loci; meQTL.
© 2023. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

References
Publication types
MeSH terms
Grants and funding
- MC_UU_00032/4/MRC_/Medical Research Council/United Kingdom
- G0000934/MRC_/Medical Research Council/United Kingdom
- MC_UU_00006/2/MRC_/Medical Research Council/United Kingdom
- BB/S020845/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/T019980/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
