

Small molecule targeting of transcription-replication conflict for selective chemotherapy
- PMID: 37531956
- PMCID: PMC10592352
- DOI: 10.1016/j.chembiol.2023.07.001
Small molecule targeting of transcription-replication conflict for selective chemotherapy
Abstract
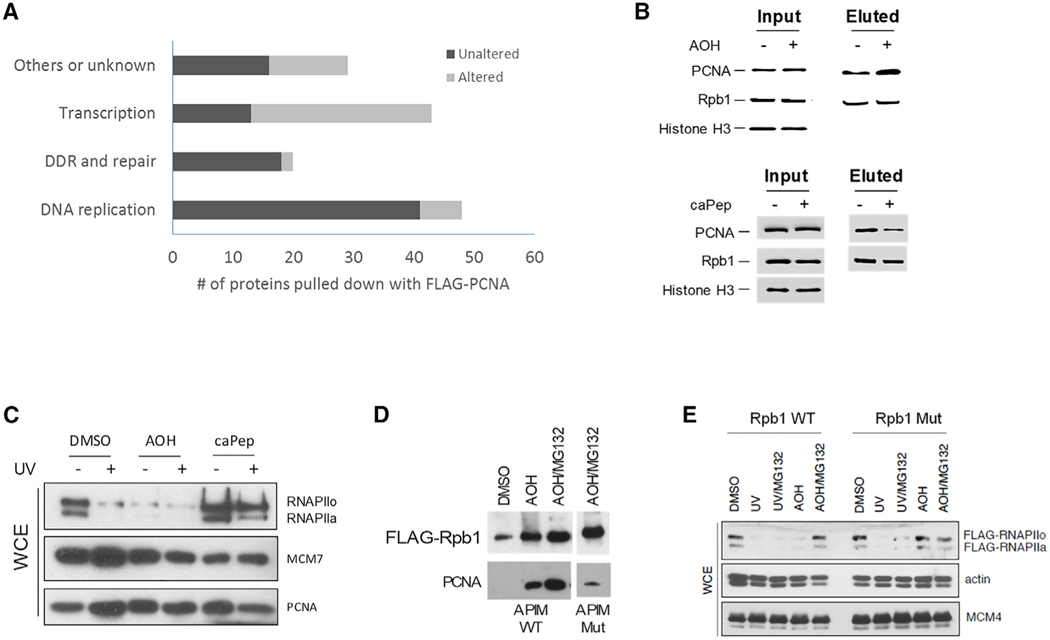
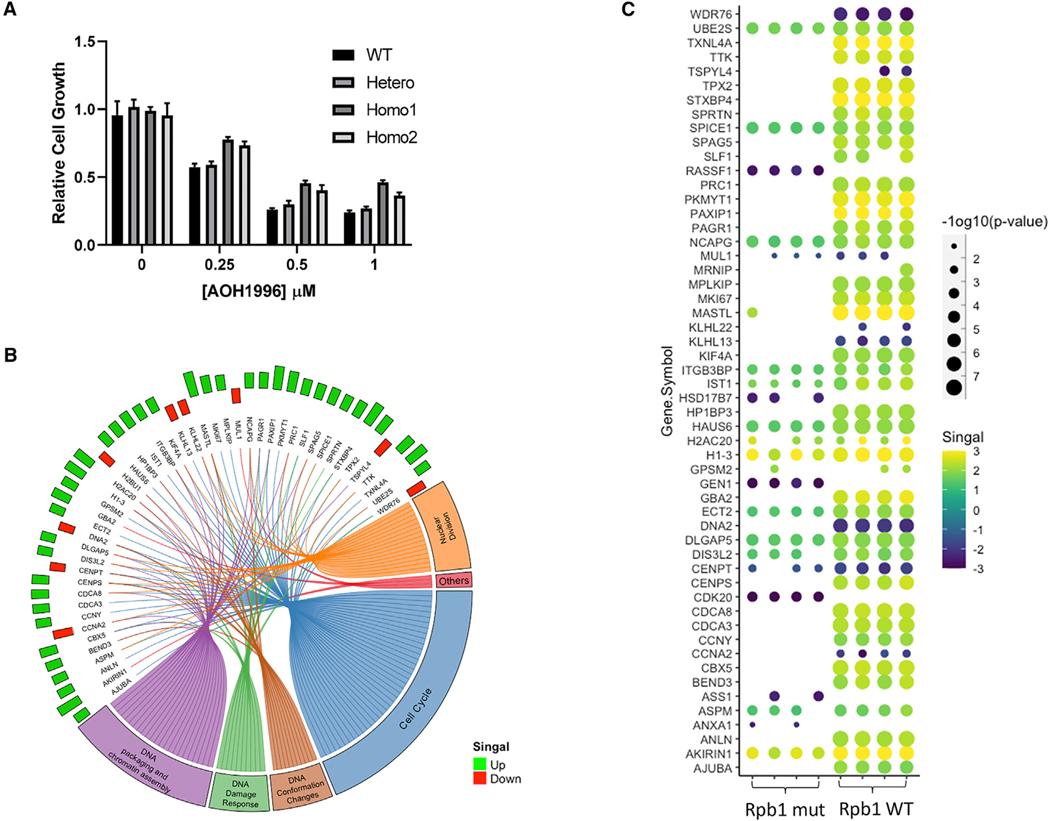
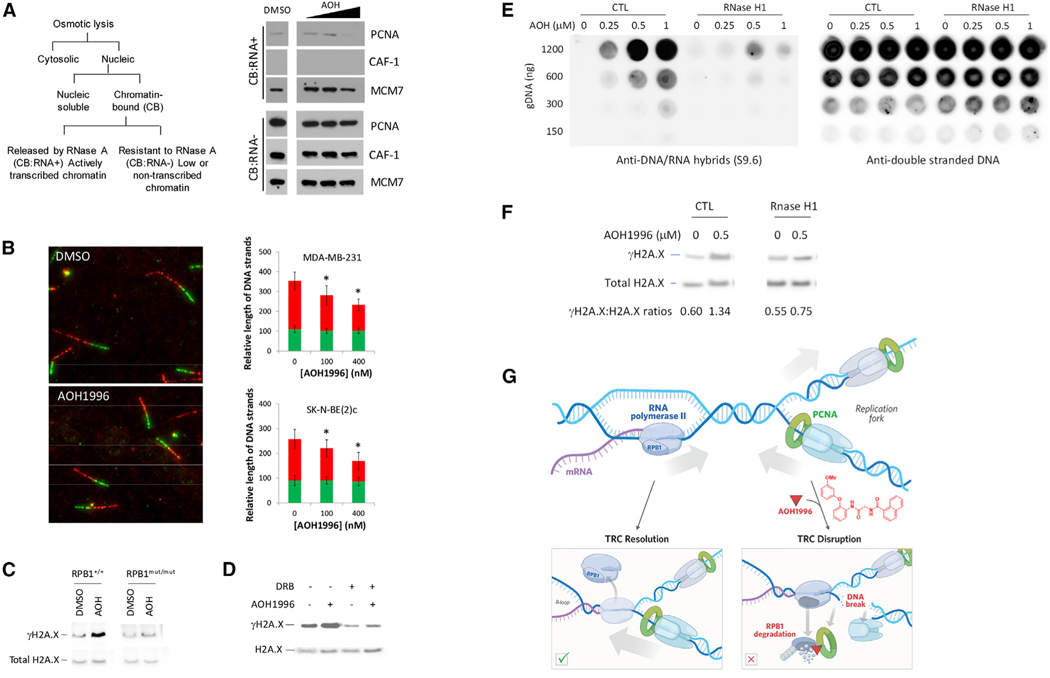
Targeting transcription replication conflicts, a major source of endogenous DNA double-stranded breaks and genomic instability could have important anticancer therapeutic implications. Proliferating cell nuclear antigen (PCNA) is critical to DNA replication and repair processes. Through a rational drug design approach, we identified a small molecule PCNA inhibitor, AOH1996, which selectively kills cancer cells. AOH1996 enhances the interaction between PCNA and the largest subunit of RNA polymerase II, RPB1, and dissociates PCNA from actively transcribed chromatin regions, while inducing DNA double-stranded breaks in a transcription-dependent manner. Attenuation of RPB1 interaction with PCNA, by a point mutation in RPB1's PCNA-binding region, confers resistance to AOH1996. Orally administrable and metabolically stable, AOH1996 suppresses tumor growth as a monotherapy or as a combination treatment but causes no discernable side effects. Inhibitors of transcription replication conflict resolution may provide a new and unique therapeutic avenue for exploiting this cancer-selective vulnerability.
Keywords: DNA repair; DNA replication stress; PCNA; transcription-replication conflict.
Copyright © 2023 The Authors. Published by Elsevier Ltd.. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare the following competing financial interest(s): City of Hope’s Office of Technology Licensing has been awarded a patent on AOH1996 and its analogs. L.H. Malkas, R.J. Hickey, D. Horne, and L. Gu are listed as inventors.
Figures
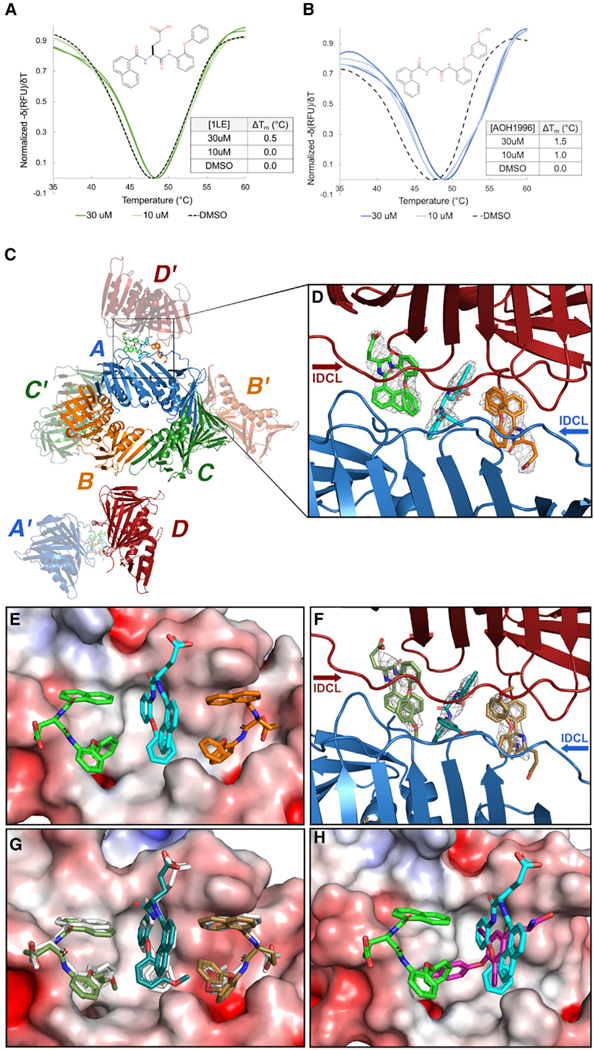
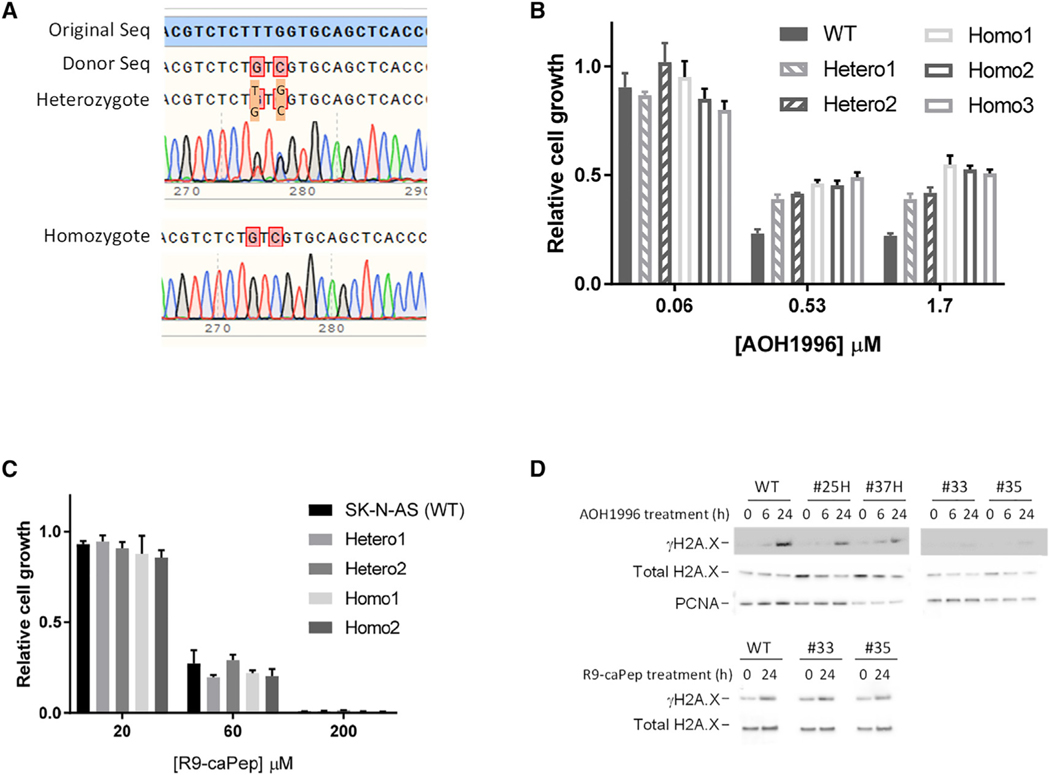
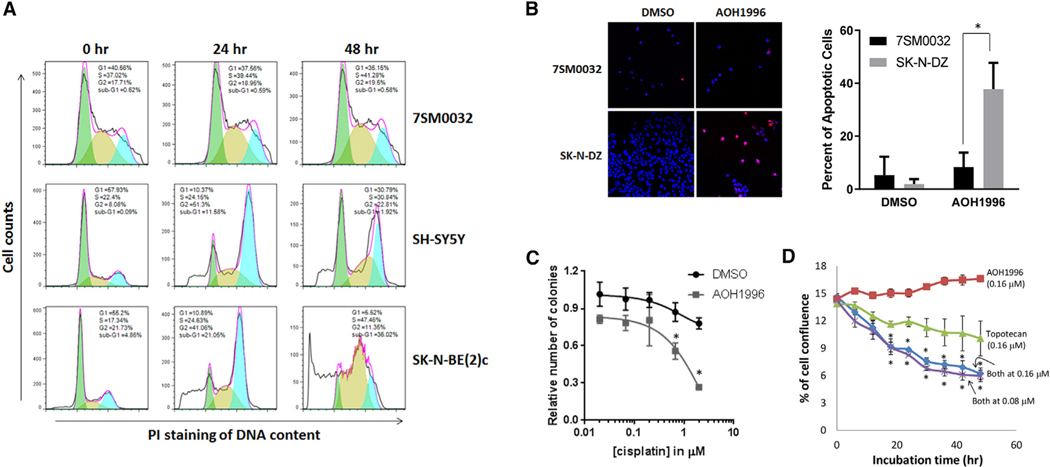
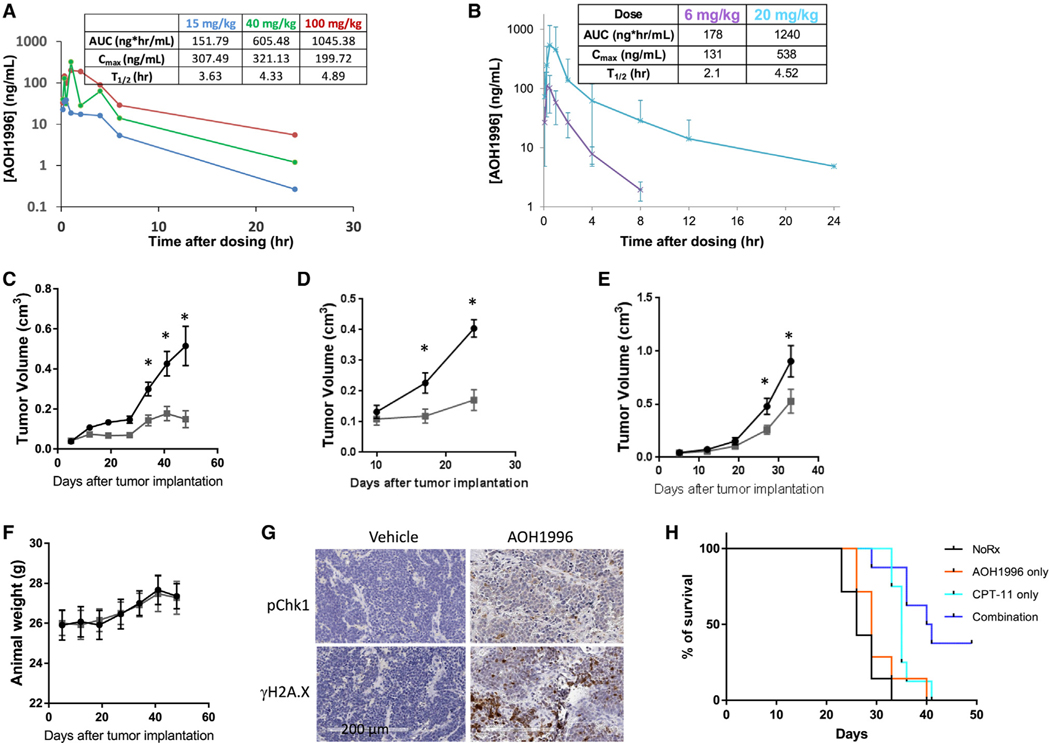
References
-
- Krishna TS, Kong XP, Gary S, Burgers PM, and Kuriyan J. (1994). Crystal structure of the eukaryotic DNA polymerase processivity factor PCNA. Cell 79, 1233–1243. - PubMed
-
- Tsutakawa SE, Van Wynsberghe AW, Freudenthal BD, Weinacht CP, Gakhar L, Washington MT, Zhuang Z, Tainer JA, and Ivanov I. (2011). Solution X-ray scattering combined with computational modeling reveals multiple conformations of covalently bound ubiquitin on PCNA. Proc. Natl. Acad. Sci. USA 108, 17672–17677. - PMC - PubMed
-
- Chapados BR, Hosfield DJ, Han S, Qiu J, Yelent B, Shen B, and Tainer JA (2004). Structural basis for FEN-1 substrate specificity and PCNA-mediated activation in DNA replication and repair. Cell 116, 39–50. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
