

Proteogenomic analysis reveals RNA as a source for tumor-agnostic neoantigen identification
- PMID: 37532709
- PMCID: PMC10397250
- DOI: 10.1038/s41467-023-39570-7
Proteogenomic analysis reveals RNA as a source for tumor-agnostic neoantigen identification
Erratum in
-
Author Correction: Proteogenomic analysis reveals RNA as a source for tumor-agnostic neoantigen identification.Nat Commun. 2024 Mar 15;15(1):2364. doi: 10.1038/s41467-024-46724-8. Nat Commun. 2024. PMID: 38491045 Free PMC article. No abstract available.
Abstract
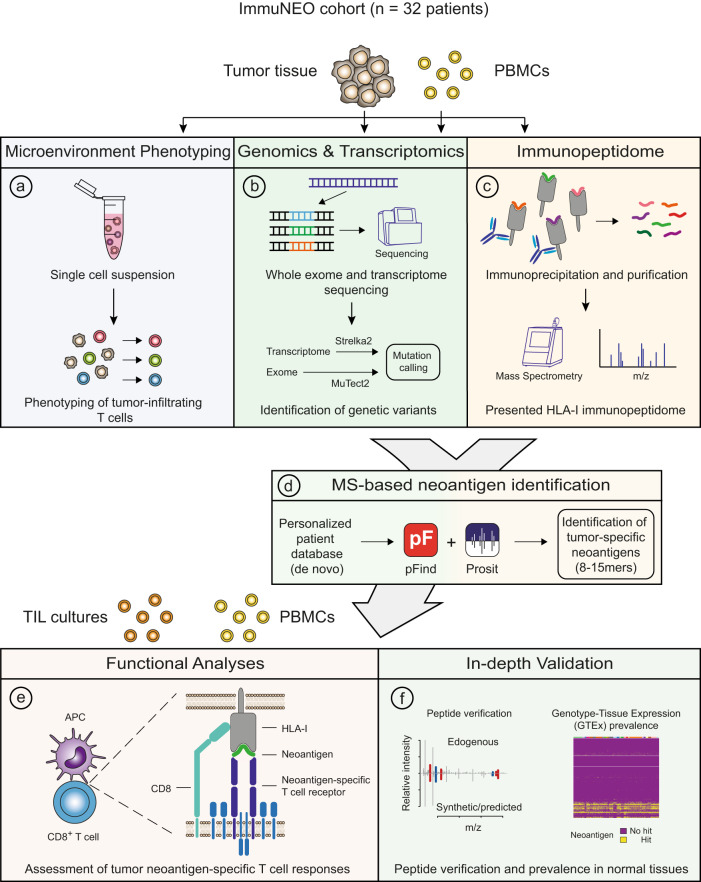
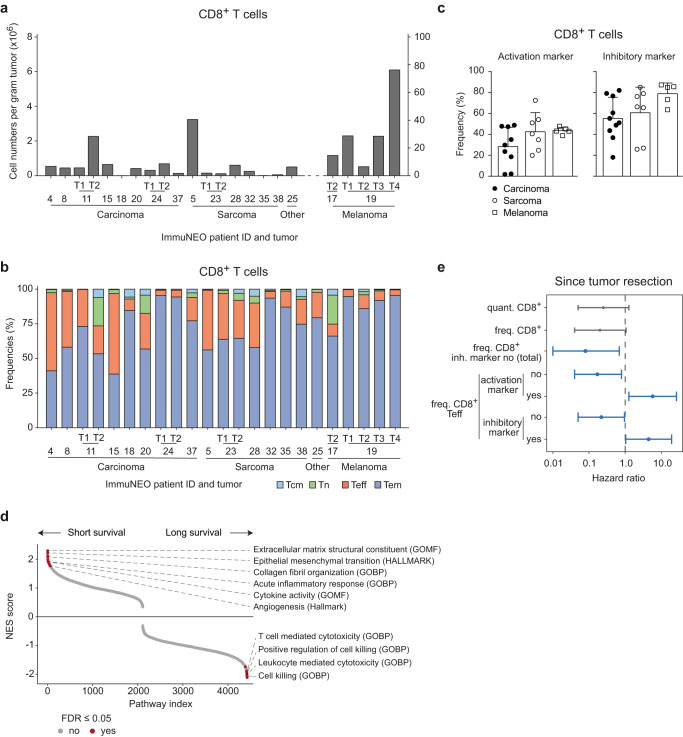
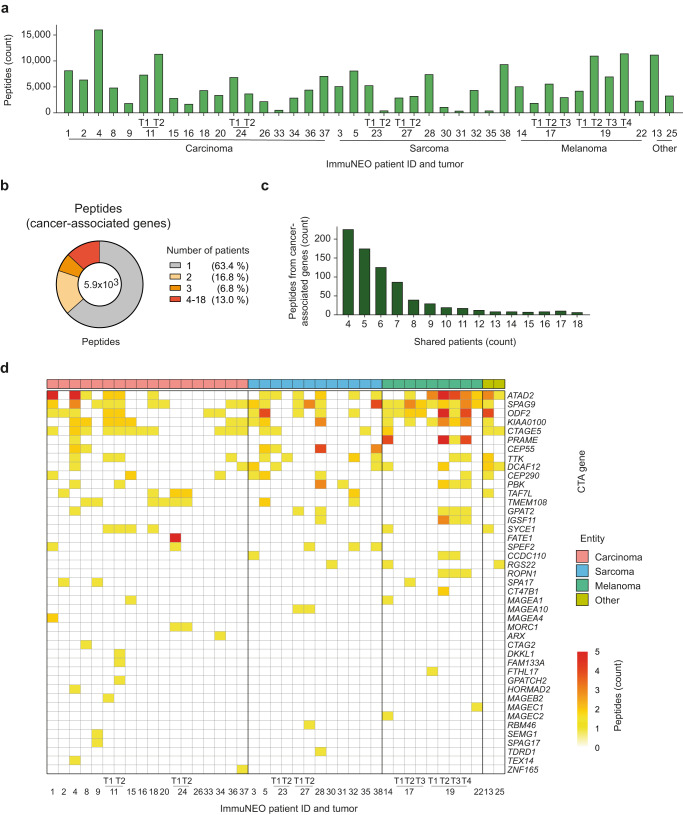
Systemic pan-tumor analyses may reveal the significance of common features implicated in cancer immunogenicity and patient survival. Here, we provide a comprehensive multi-omics data set for 32 patients across 25 tumor types for proteogenomic-based discovery of neoantigens. By using an optimized computational approach, we discover a large number of tumor-specific and tumor-associated antigens. To create a pipeline for the identification of neoantigens in our cohort, we combine DNA and RNA sequencing with MS-based immunopeptidomics of tumor specimens, followed by the assessment of their immunogenicity and an in-depth validation process. We detect a broad variety of non-canonical HLA-binding peptides in the majority of patients demonstrating partially immunogenicity. Our validation process allows for the selection of 32 potential neoantigen candidates. The majority of neoantigen candidates originates from variants identified in the RNA data set, illustrating the relevance of RNA as a still understudied source of cancer antigens. This study underlines the importance of RNA-centered variant detection for the identification of shared biomarkers and potentially relevant neoantigen candidates.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures




References
-
- Bräunlein, E. et al. Spatial and temporal plasticity of neoantigen-specific T-cell responses bases on characteristics associated to antigen and TCR. Preprint at bioRxiv10.1101/2021.02.02.428777 (2021).
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
