

Protocol for the development of coarse-grained structures for macromolecular simulation using GROMACS
- PMID: 37535543
- PMCID: PMC10399882
- DOI: 10.1371/journal.pone.0288264
Protocol for the development of coarse-grained structures for macromolecular simulation using GROMACS
Abstract
Coarse-grained simulations have emerged as a valuable tool in the study of large and complex biomolecular systems. These simulations, which use simplified models to represent complex biomolecules, reduce the computational cost of simulations and enable the study of larger systems for longer periods of time than traditional atomistic simulations. GROMACS is a widely used software package for performing coarse-grained simulations of biomolecules, and several force fields have been developed specifically for this purpose. In this protocol paper, we explore the advantages of using coarse-grained simulations in the study of biomolecular systems, focusing specifically on simulations performed using GROMACS. We discuss the force fields required for these simulations and the types of research questions that can be addressed using coarse-grained simulations. We also highlight the potential benefits of coarse-grained simulations for the development of new force fields and simulation methodologies. We then discuss the expected results from coarse-grained simulations using GROMACS and the various techniques that can be used to analyze these results. We explore the use of trajectory analysis tools, as well as thermodynamic and structural analysis techniques, to gain insight into the behavior of biomolecular systems.
Copyright: © 2023 Niranjan et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
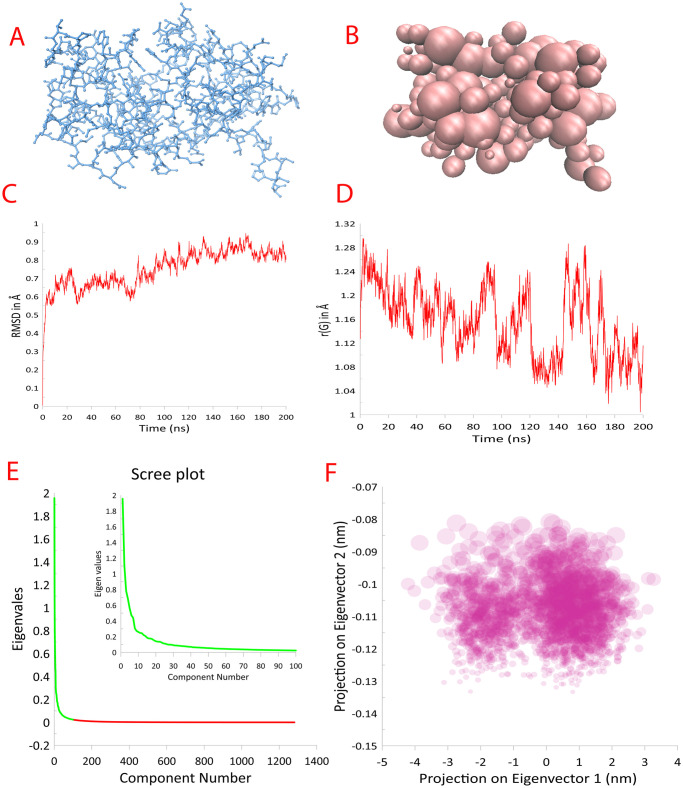
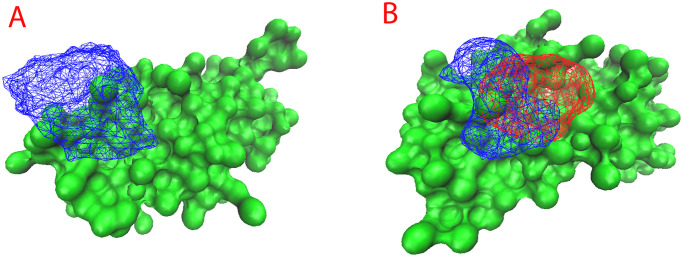
References
-
- Peter C., & Kremer K. (2009). Multiscale simulation of soft matter systems. Faraday Discussions, 144, 9–24. - PubMed
-
- Abraham M. J., Murtola T., Schulz R., Páll S., Smith J. C., Hess B., et al. (2015). Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX, 1–2, 19–25. doi: 10.1016/j.softx.2015.06.001 - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
