

The dawn of spatial omics
- PMID: 37535749
- PMCID: PMC7614974
- DOI: 10.1126/science.abq4964
The dawn of spatial omics
Abstract
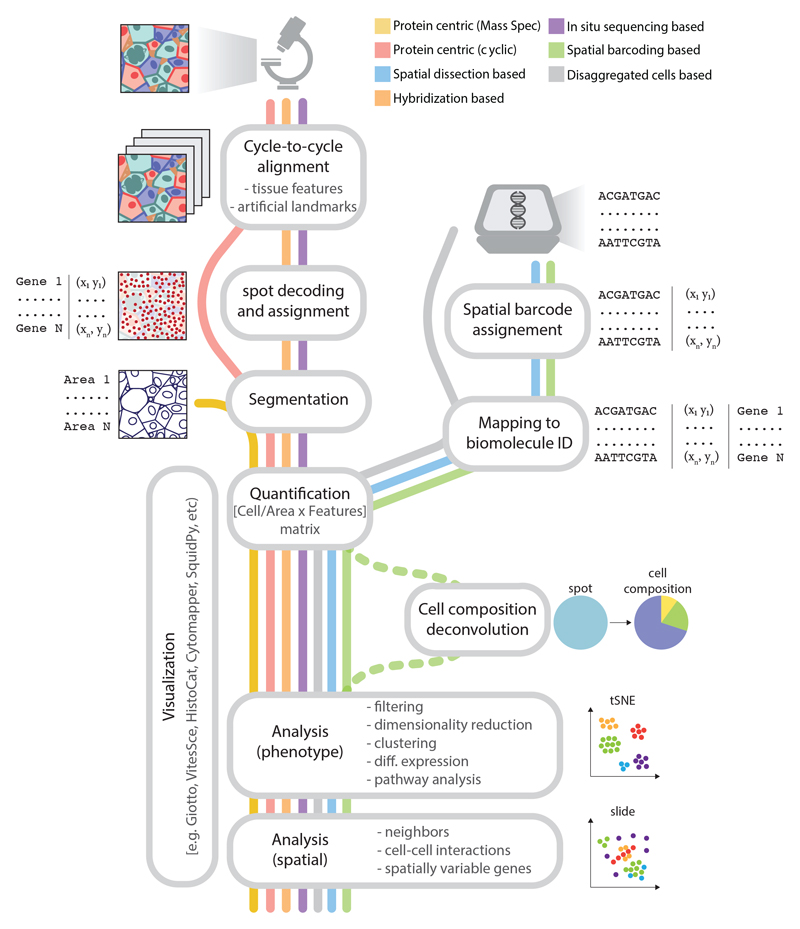
Spatial omics has been widely heralded as the new frontier in life sciences. This term encompasses a wide range of techniques that promise to transform many areas of biology and eventually revolutionize pathology by measuring physical tissue structure and molecular characteristics at the same time. Although the field came of age in the past 5 years, it still suffers from some growing pains: barriers to entry, robustness, unclear best practices for experimental design and analysis, and lack of standardization. In this Review, we present a systematic catalog of the different families of spatial omics technologies; highlight their principles, power, and limitations; and give some perspective and suggestions on the biggest challenges that lay ahead in this incredibly powerful-but still hard to navigate-landscape.
Conflict of interest statement
D.B. and G.J.H. are cofounders of Suil Interactive, a company focused on the development of virtual reality tools for the visualization of spatial profiling data, and of Elyx, a company developing and commercializing spatial profiling technologies. G.B. is a shareholder of Elyx. D.B., G.J.H., and G.B. are listed as inventors on patent applications relating to the spatial profiling field, including WO2021105723A1 and WO2021116715A1
Figures

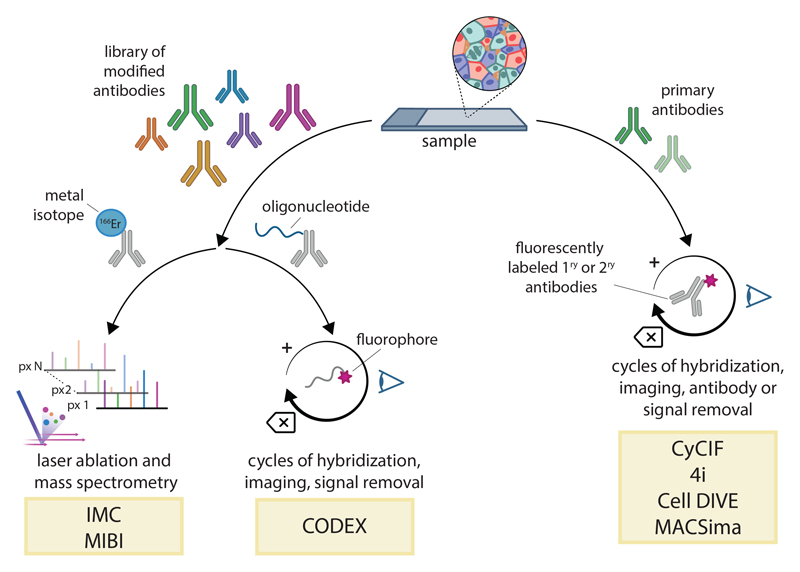
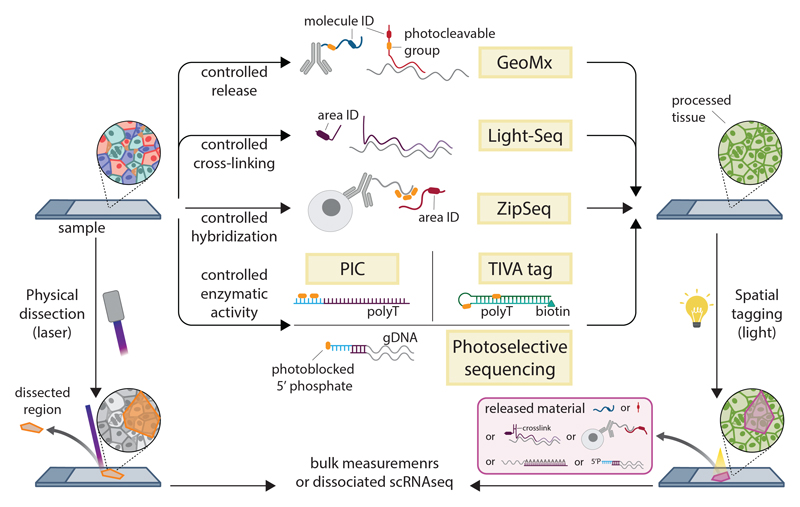
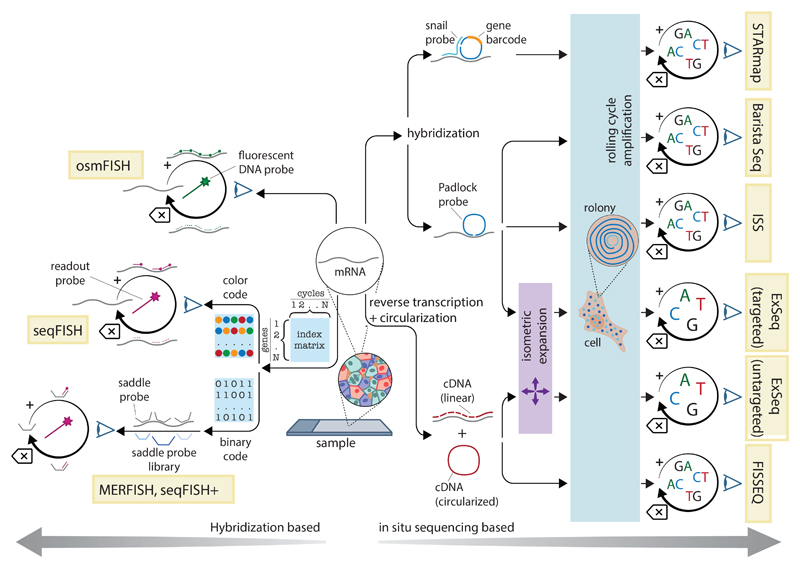
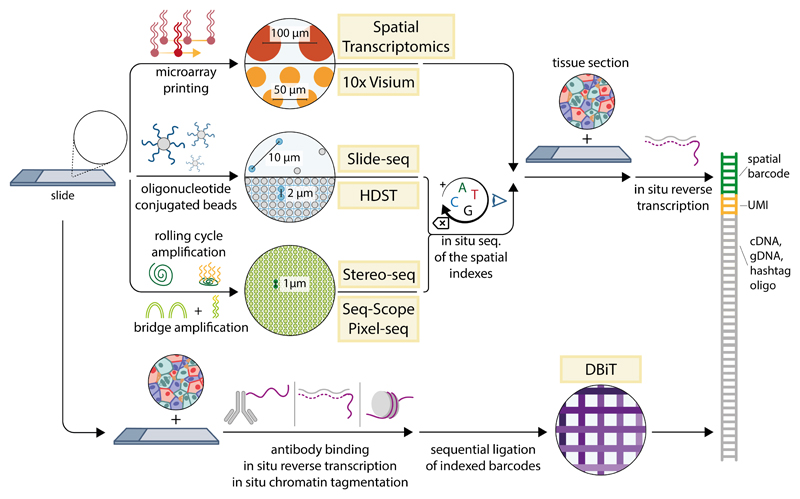
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
