

Cancer cell plasticity during tumor progression, metastasis and response to therapy
- PMID: 37537300
- PMCID: PMC7615147
- DOI: 10.1038/s43018-023-00595-y
Cancer cell plasticity during tumor progression, metastasis and response to therapy
Abstract
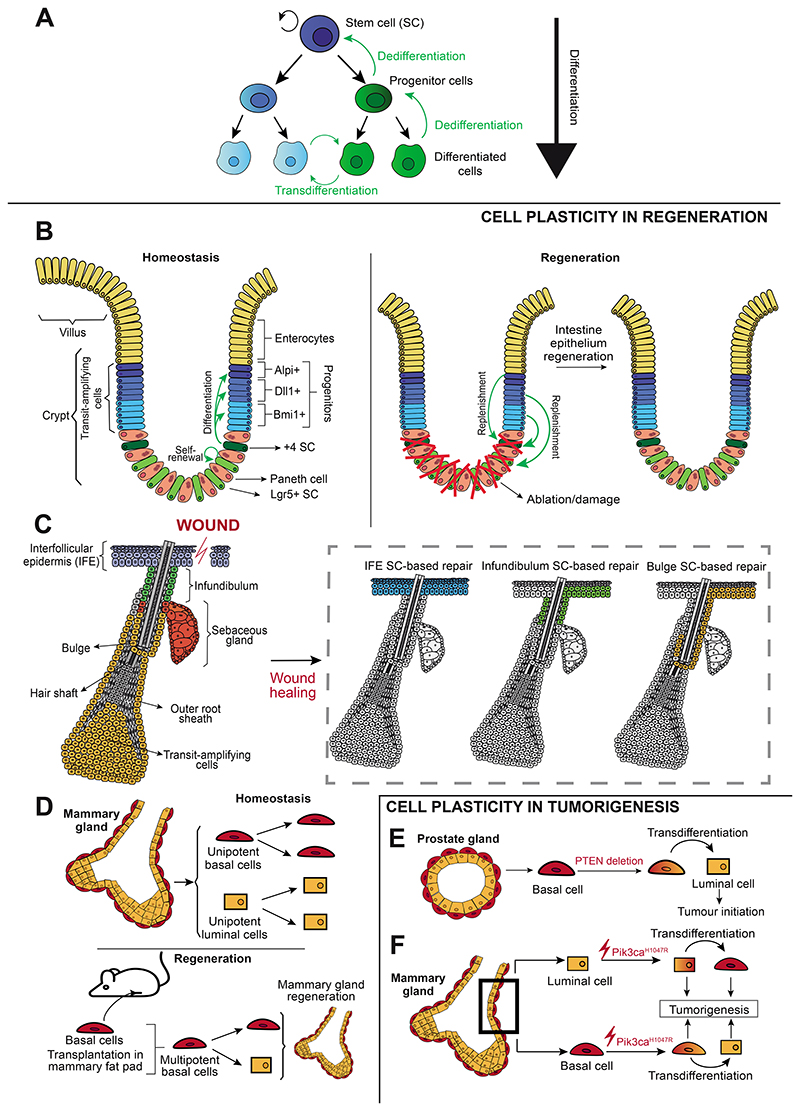
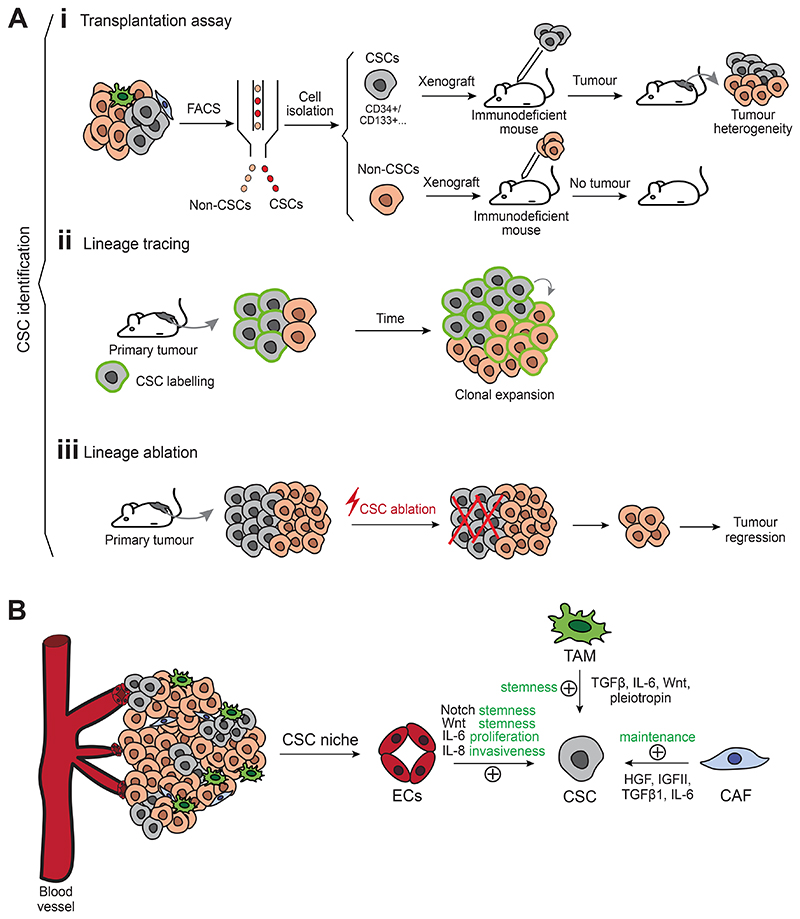
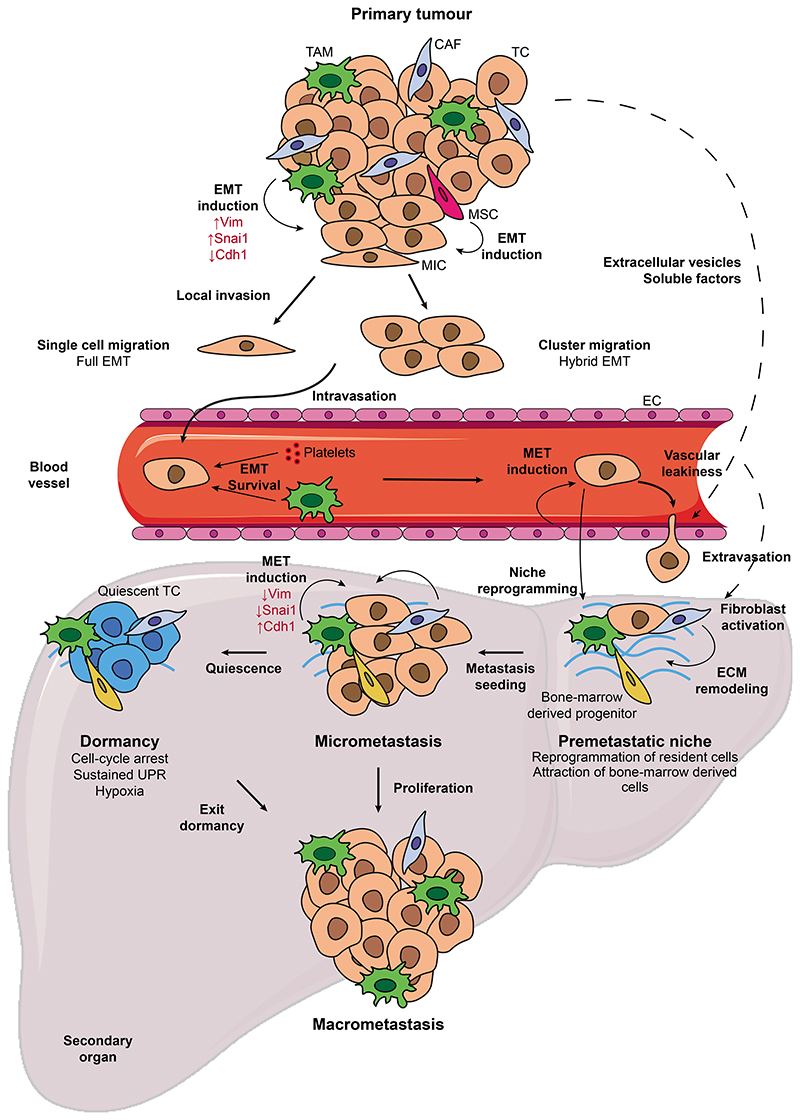
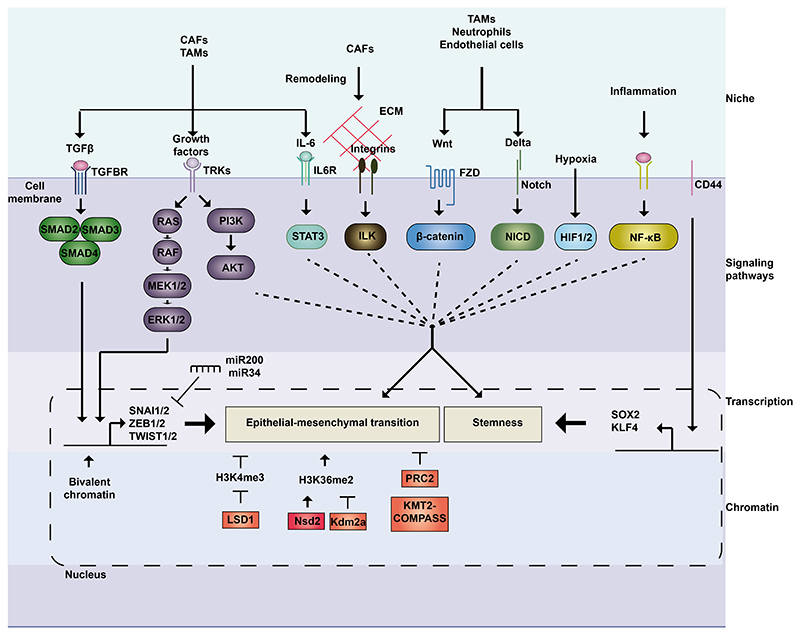
Cell plasticity represents the ability of cells to be reprogrammed and to change their fate and identity, enabling homeostasis restoration and tissue regeneration following damage. Cell plasticity also contributes to pathological conditions, such as cancer, enabling cells to acquire new phenotypic and functional features by transiting across distinct cell states that contribute to tumor initiation, progression, metastasis and resistance to therapy. Here, we review the intrinsic and extrinsic mechanisms driving cell plasticity that promote tumor growth and proliferation as well as metastasis and drug tolerance. Finally, we discuss how cell plasticity could be exploited for anti-cancer therapy.
© 2023. Springer Nature America, Inc.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures

References
-
- Gurdon JB. The Developmental Capacity of Nuclei taken from Intestinal Epithelium Cells of Feeding Tadpoles. Development. 1962;10:622–640. - PubMed
-
- Takahashi K, Yamanaka S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell. 2006;126:663–676. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
