

Systematic benchmarking of single-cell ATAC-sequencing protocols
- PMID: 37537502
- PMCID: PMC11180611
- DOI: 10.1038/s41587-023-01881-x
Systematic benchmarking of single-cell ATAC-sequencing protocols
Abstract
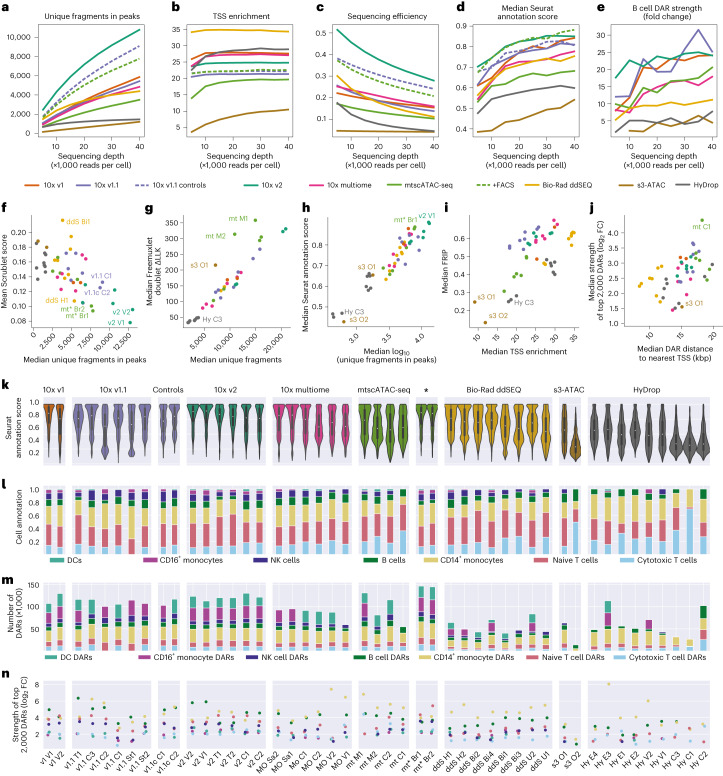
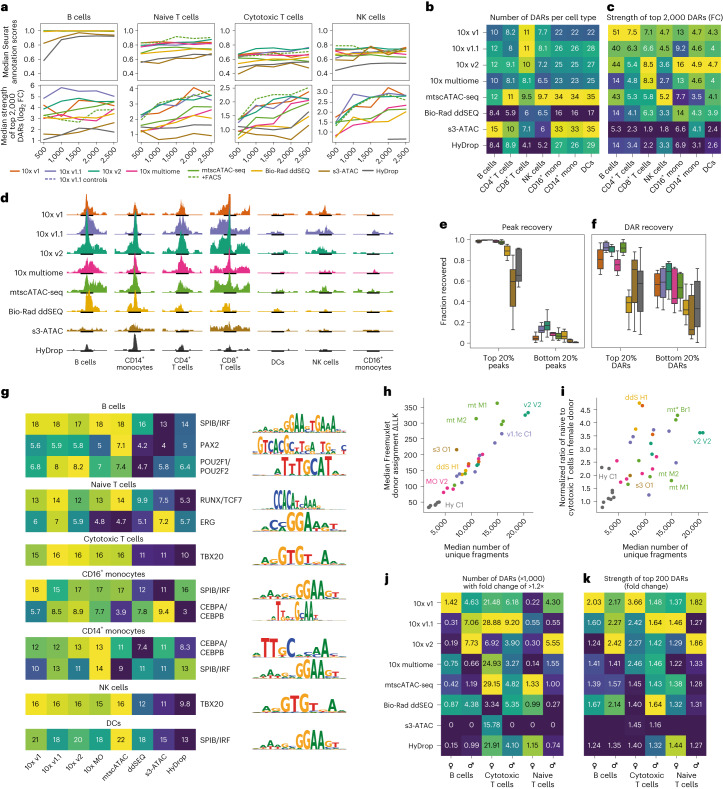
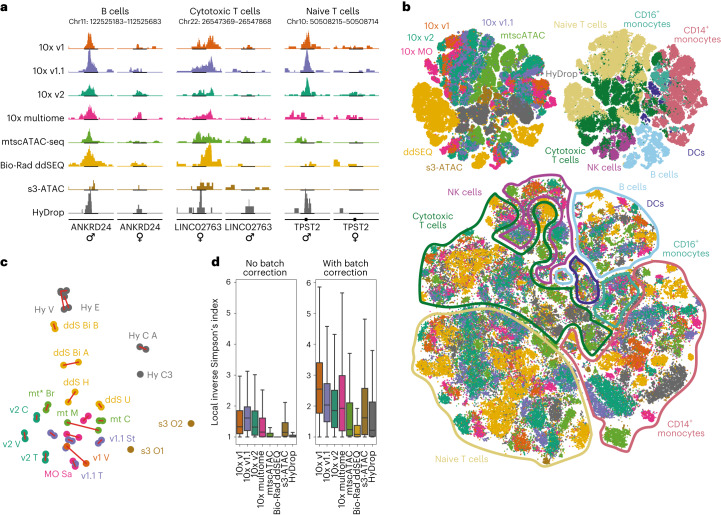
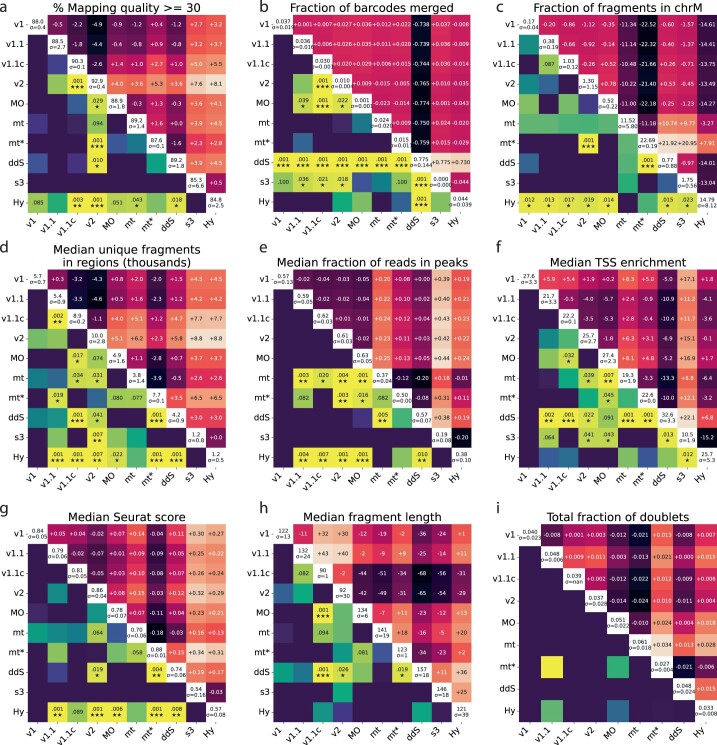
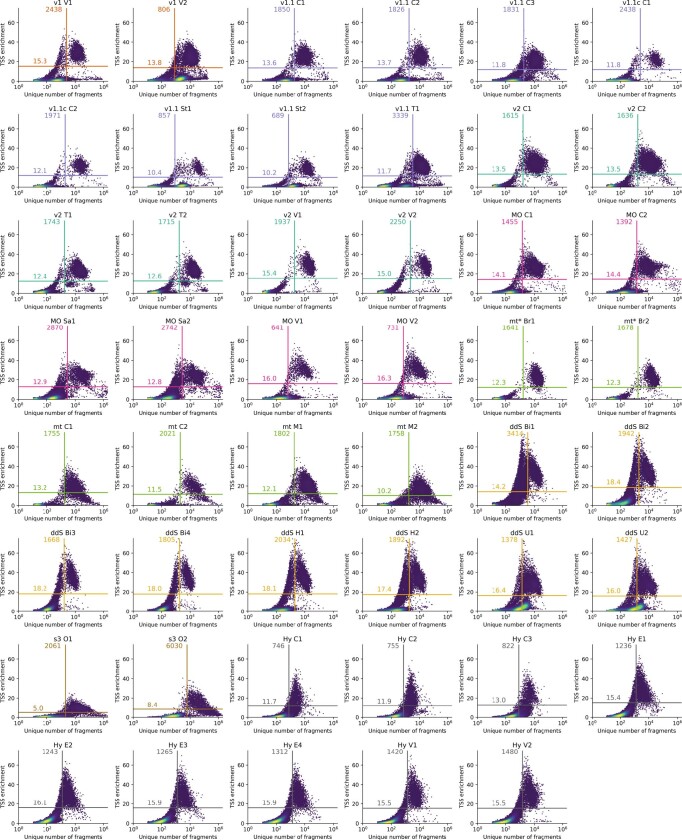
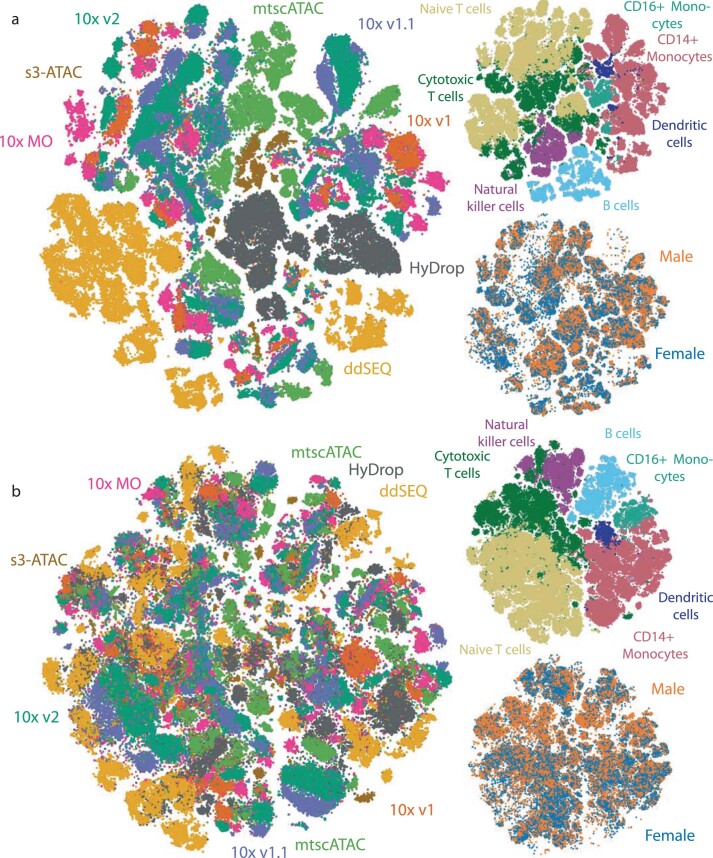
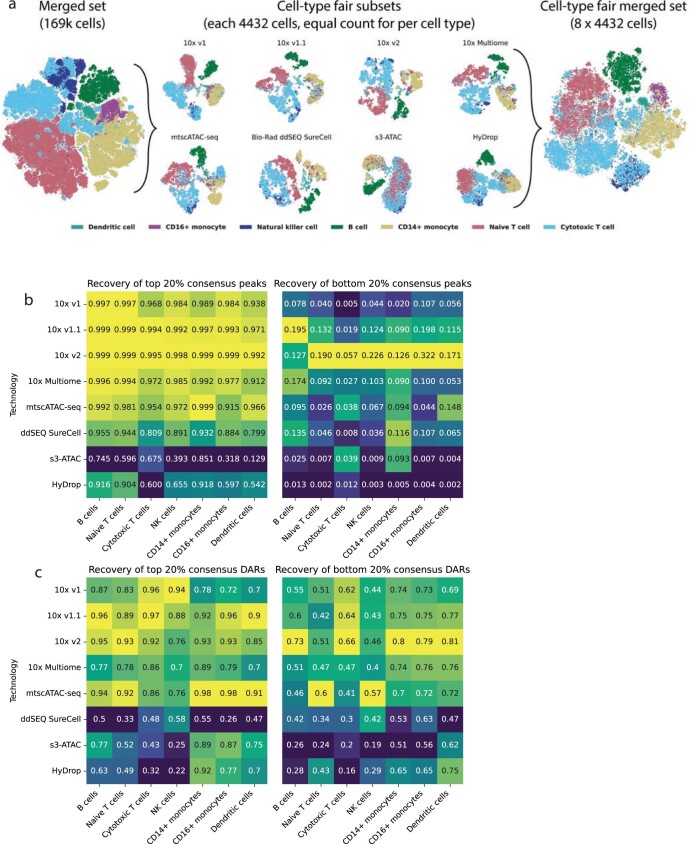
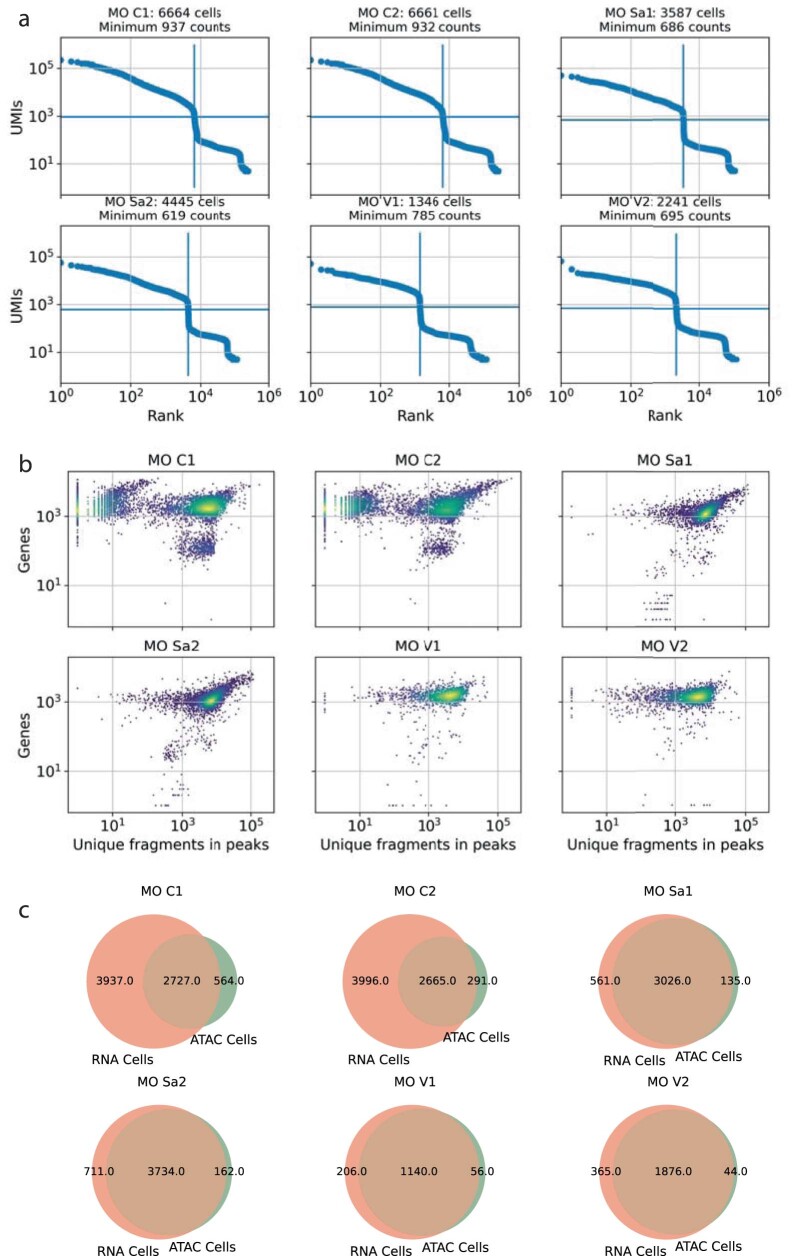
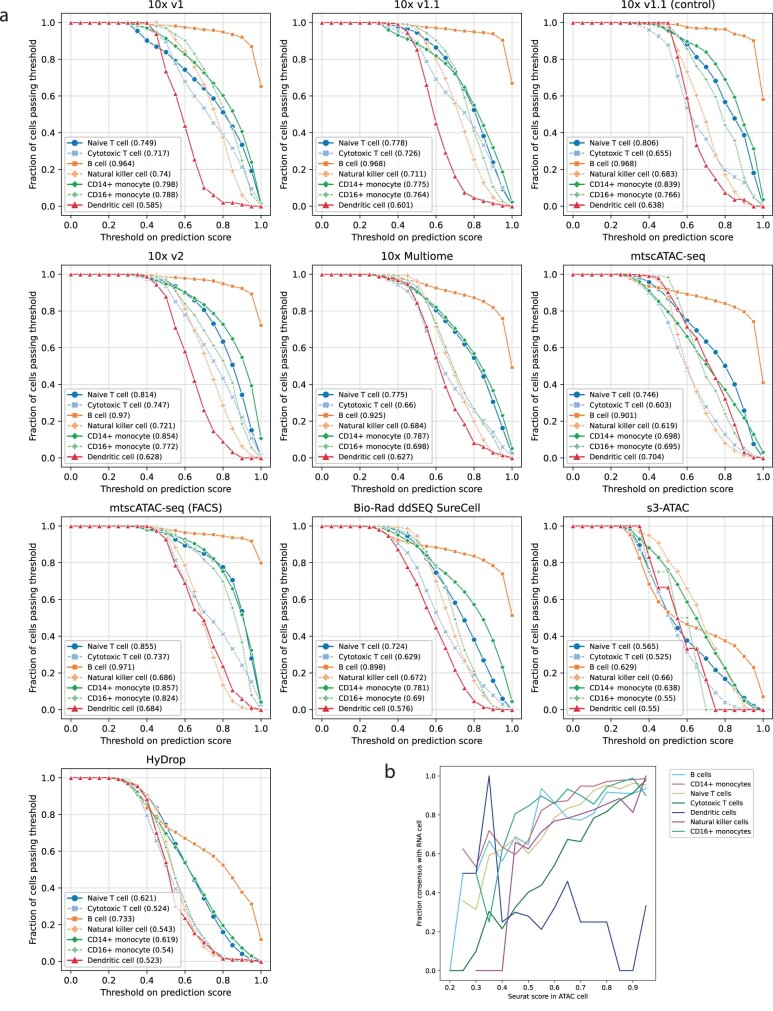
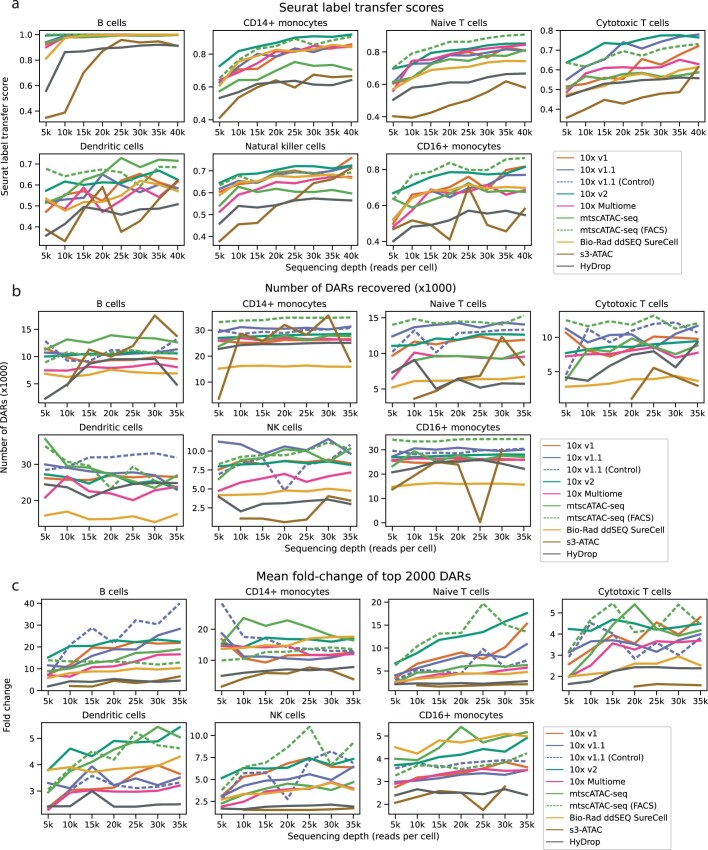
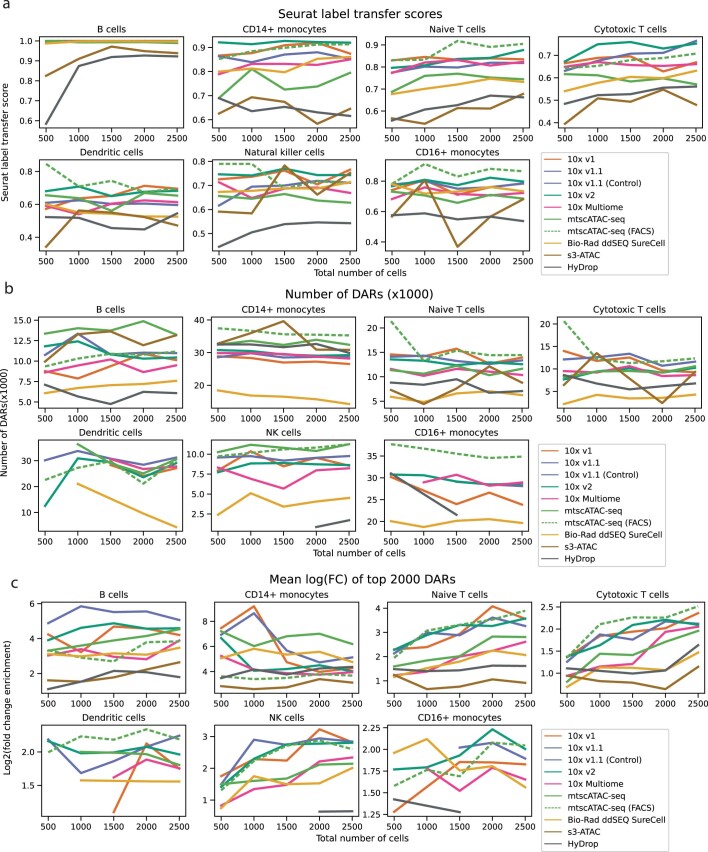
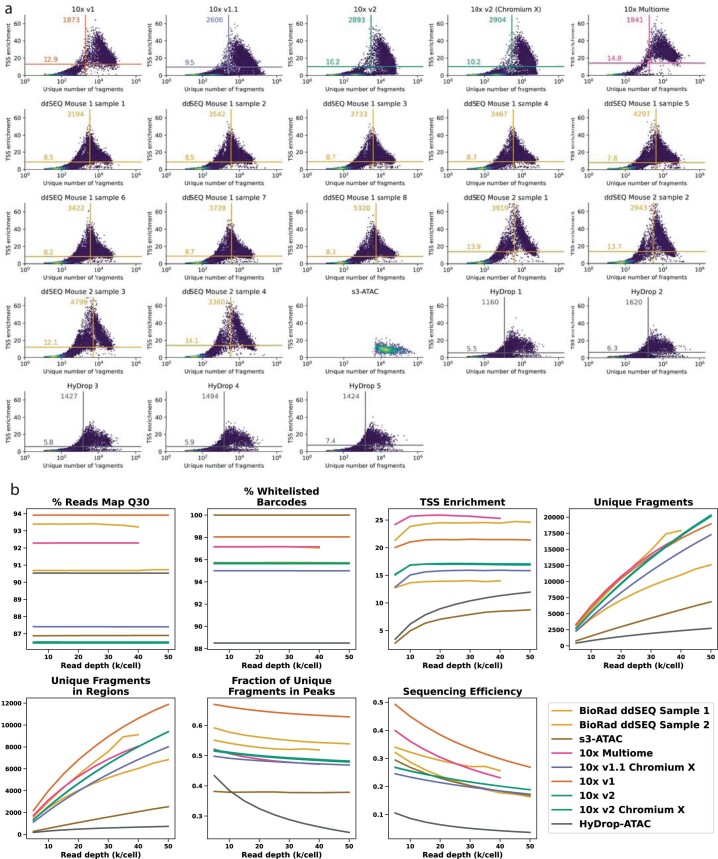
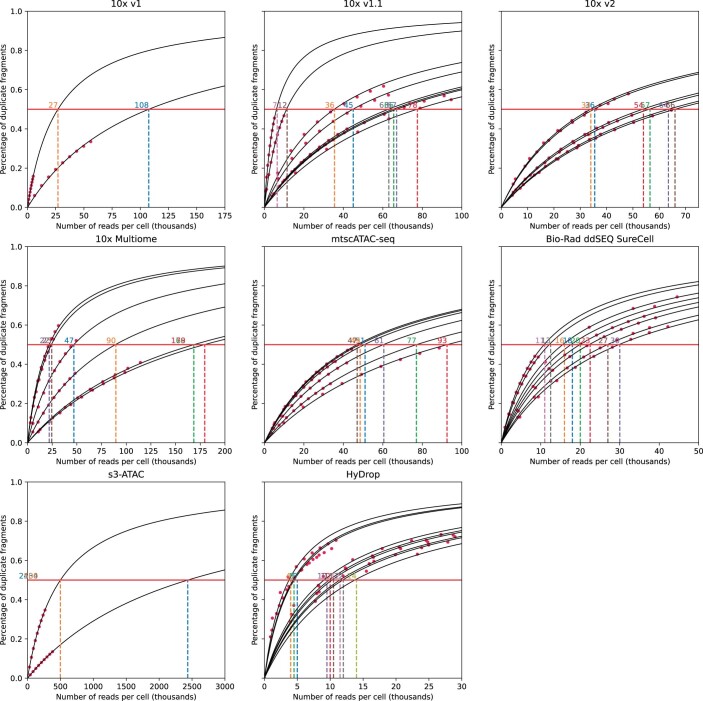
Single-cell assay for transposase-accessible chromatin by sequencing (scATAC-seq) has emerged as a powerful tool for dissecting regulatory landscapes and cellular heterogeneity. However, an exploration of systemic biases among scATAC-seq technologies has remained absent. In this study, we benchmark the performance of eight scATAC-seq methods across 47 experiments using human peripheral blood mononuclear cells (PBMCs) as a reference sample and develop PUMATAC, a universal preprocessing pipeline, to handle the various sequencing data formats. Our analyses reveal significant differences in sequencing library complexity and tagmentation specificity, which impact cell-type annotation, genotype demultiplexing, peak calling, differential region accessibility and transcription factor motif enrichment. Our findings underscore the importance of sample extraction, method selection, data processing and total cost of experiments, offering valuable guidance for future research. Finally, our data and analysis pipeline encompasses 169,000 PBMC scATAC-seq profiles and a best practices code repository for scATAC-seq data analysis, which are freely available to extend this benchmarking effort to future protocols.
© 2023. The Author(s).
Conflict of interest statement
H.H. is a cofounder and equity holder of Omniscope, a Scientific Advisory Board member of MiRXES and a consultant to Moderna. A.R. is a founder and equity holder of Celsius Therapeutics, an equity holder in Immunitas Therapeutics and, until 31 August 2020, was a Scientific Advisory Board member of Syros Pharmaceuticals, Neogene Therapeutics, Asimov and Thermo Fisher Scientific. A.R. is an employee of Genentech and has equity in Roche. O.R.-R. is an employee of Genentech. A.R. and O.R.-R. are co-inventors on patent applications filed by the Broad Institute relating to single-cell genomics. In the past 3 years, S.A.T. has received remuneration for Scientific Advisory Board Membership from Sanofi, GlaxoSmithKline, Foresite Labs and Qiagen. S.A.T. is a cofounder of and holds equity in Transition Bio. 10x Genomics has licensed intellectual property on which W.J.G. is listed as an inventor. W.J.G. holds options in 10x Genomics and is a consultant for Ultima Genomics, Guardant Health. W.J.G. is a scientific cofounder of Protillion Biosciences. J.D.B. holds patents related to ATAC-seq and SHARE-seq and is a Scientific Advisory Board member of Camp4 and seqWell. A.A. is an author on licensed patents that cover the nucleosome disruption and indexed tagmentation design. L.L. holds a patent for lineage tracing using mitochondrial genome mutations and single-cell genomics and is a consultant to Cartography Biosciences. B.D. is cofounder and shareholder of Alithea Genomics. The remaining authors declare no competing interests.
Figures

References
MeSH terms
Substances
Grants and funding
- R01 DA047237/DA/NIDA NIH HHS/United States
- R00 AG059918/AG/NIA NIH HHS/United States
- U19 AI057266/AI/NIAID NIH HHS/United States
- G0B5619N/Fonds Wetenschappelijk Onderzoek (Research Foundation Flanders)
- RF1 MH128842/MH/NIMH NIH HHS/United States
- UM1 HG009436/HG/NHGRI NIH HHS/United States
- 1S80920N/Fonds Wetenschappelijk Onderzoek (Research Foundation Flanders)
- UM1 HG012076/HG/NHGRI NIH HHS/United States
- RM1 HG007735/HG/NHGRI NIH HHS/United States
- G094121N/Fonds Wetenschappelijk Onderzoek (Research Foundation Flanders)
- R35 GM124704/GM/NIGMS NIH HHS/United States
- UM1 HG009442/HG/NHGRI NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
