

Topography of mutational signatures in human cancer
- PMID: 37540596
- PMCID: PMC10507738
- DOI: 10.1016/j.celrep.2023.112930
Topography of mutational signatures in human cancer
Abstract
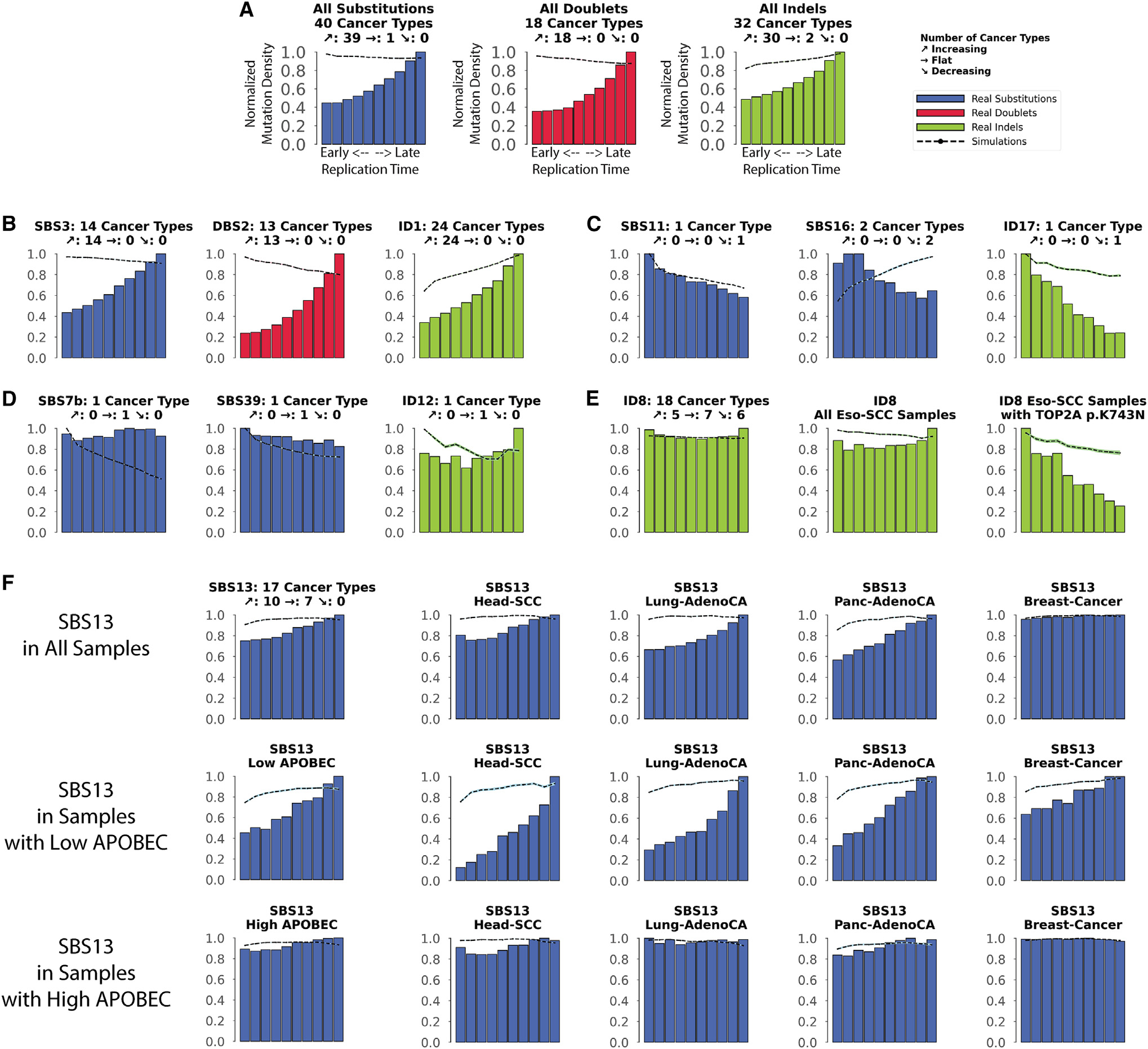
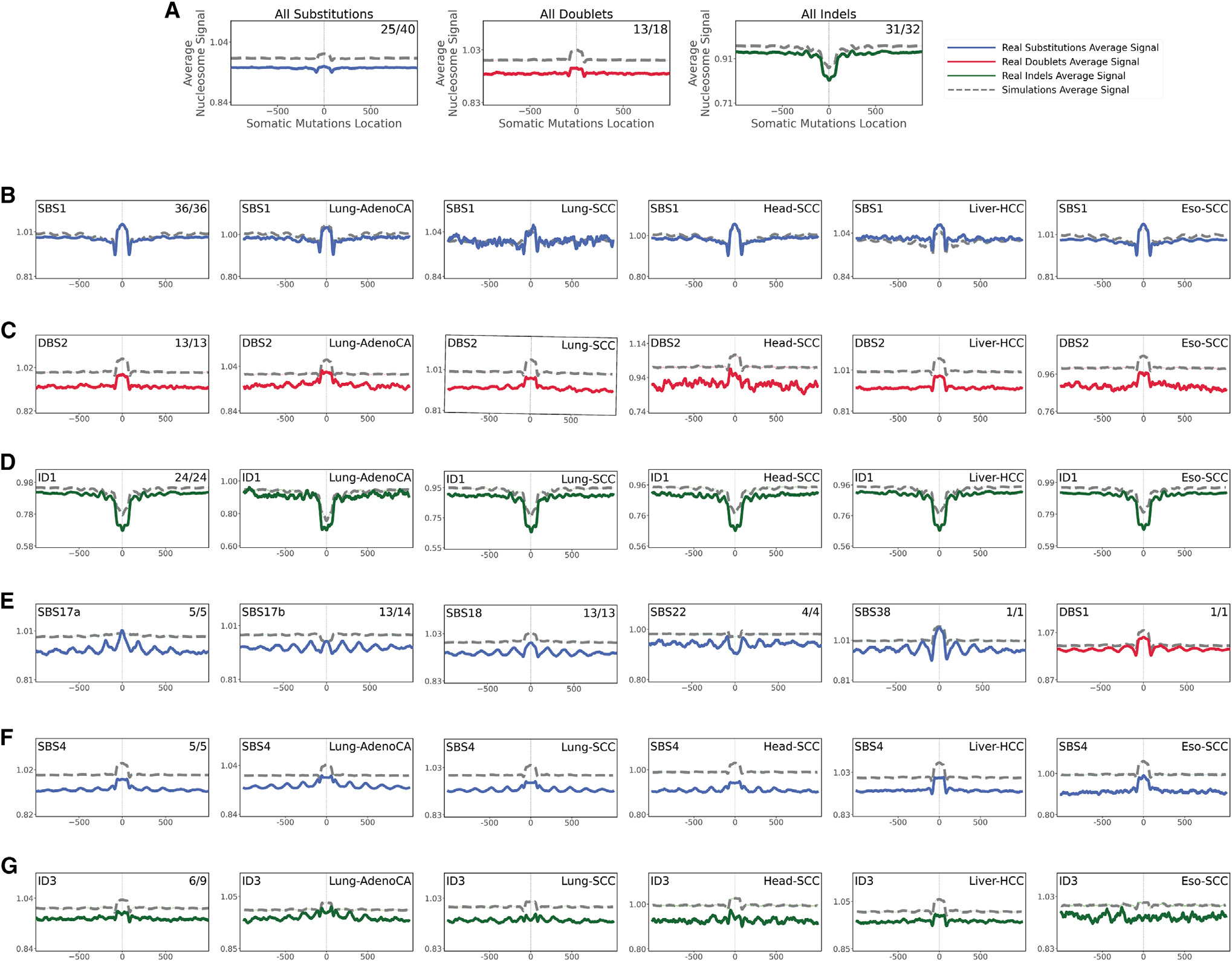
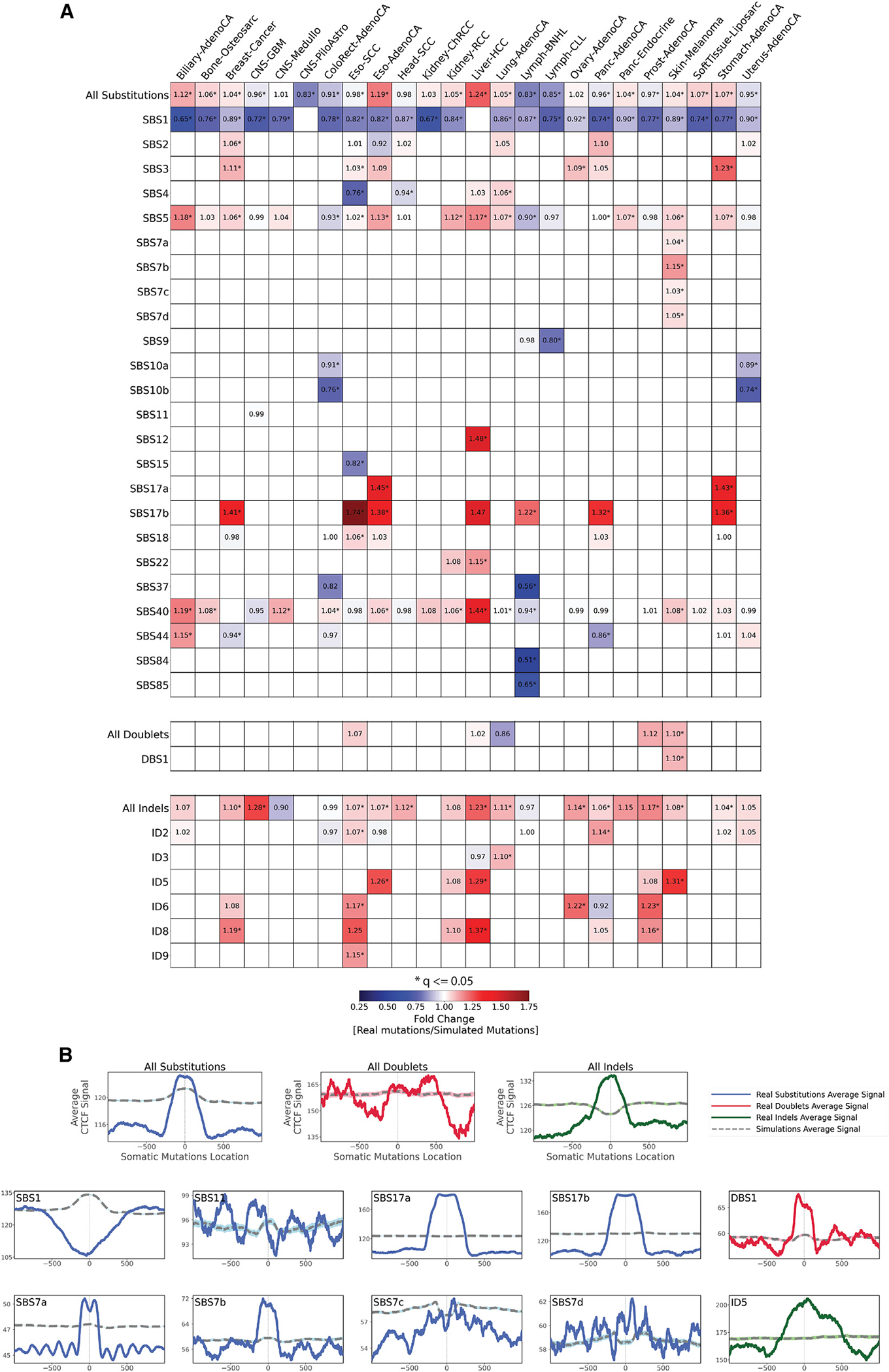
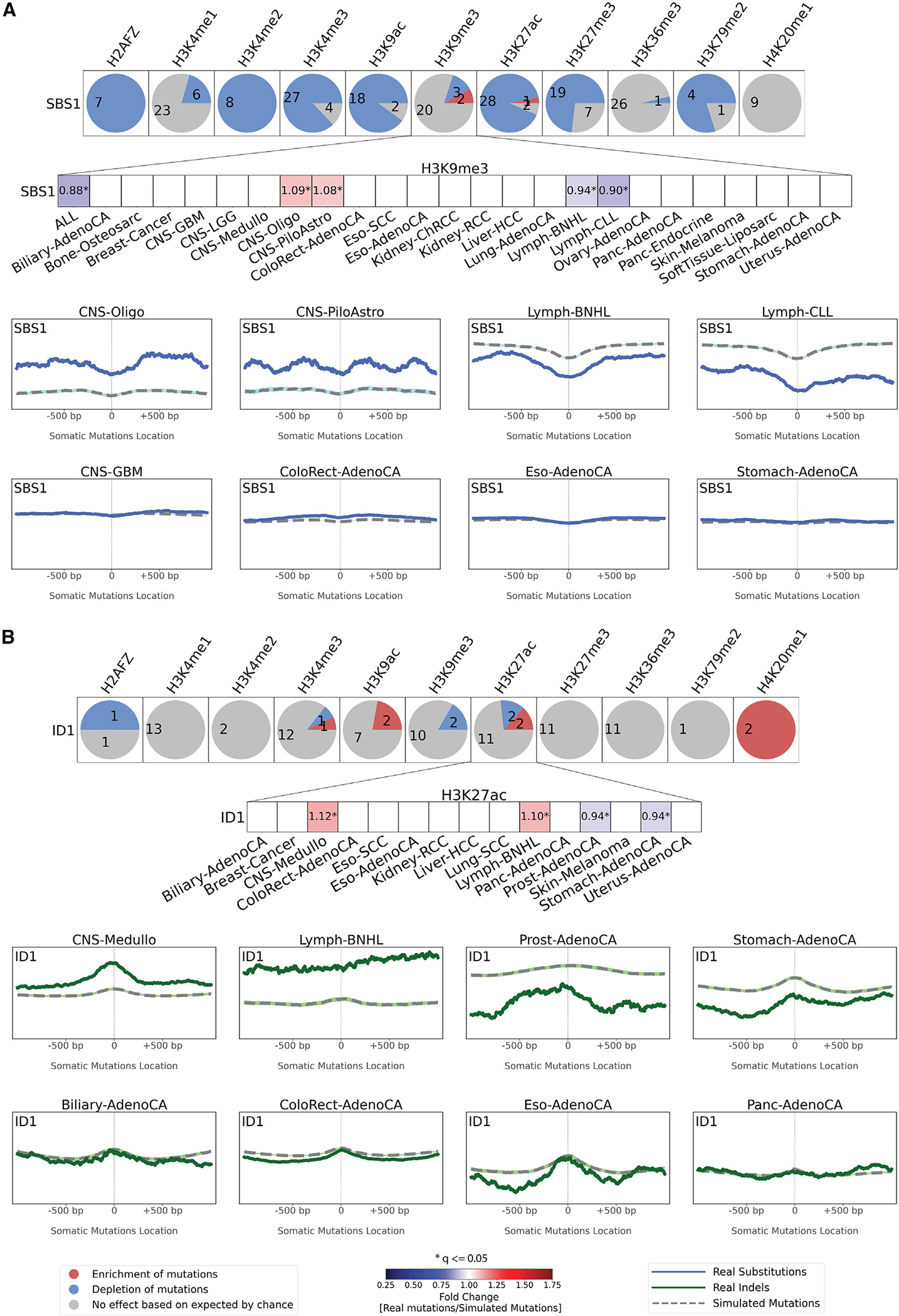
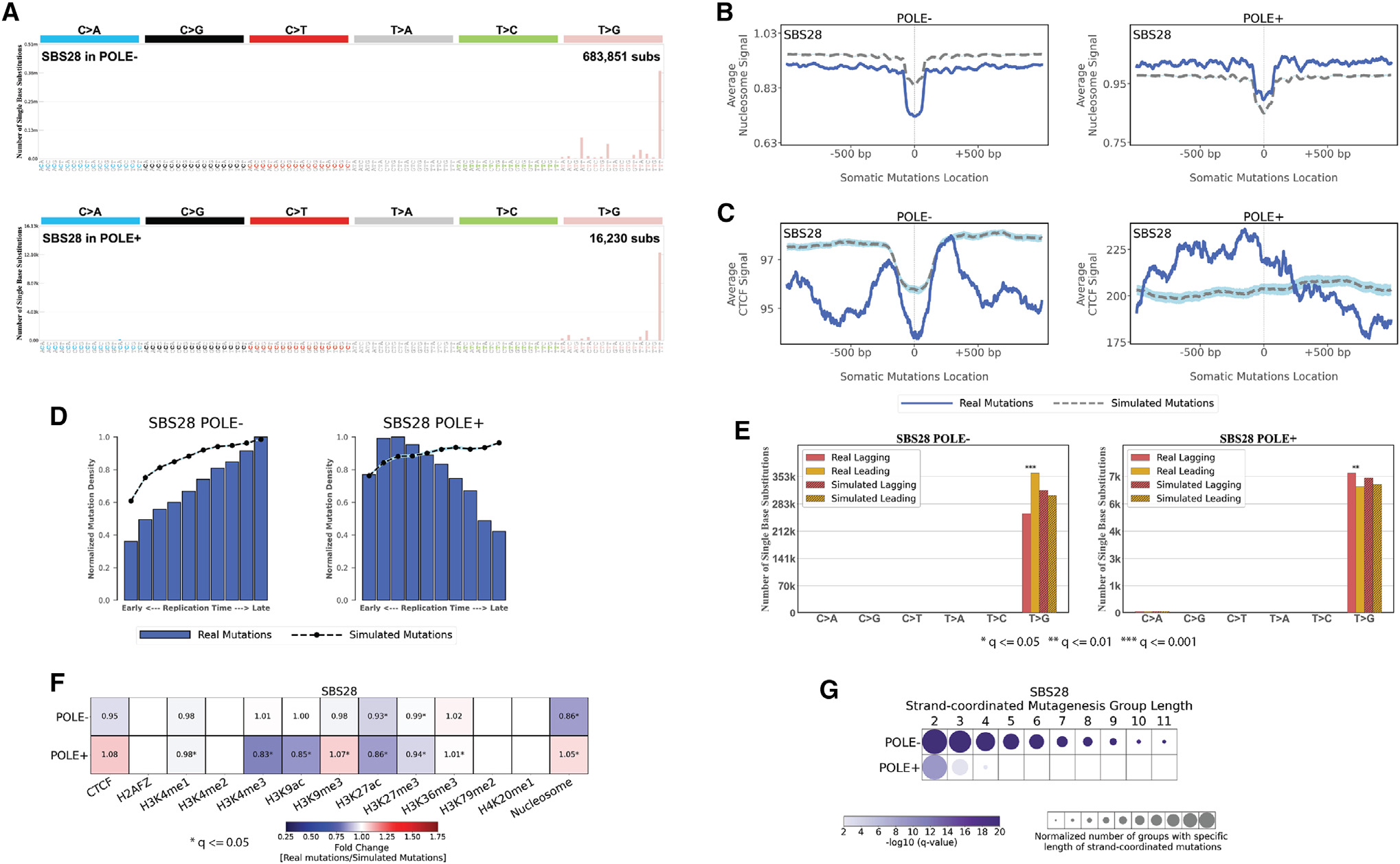
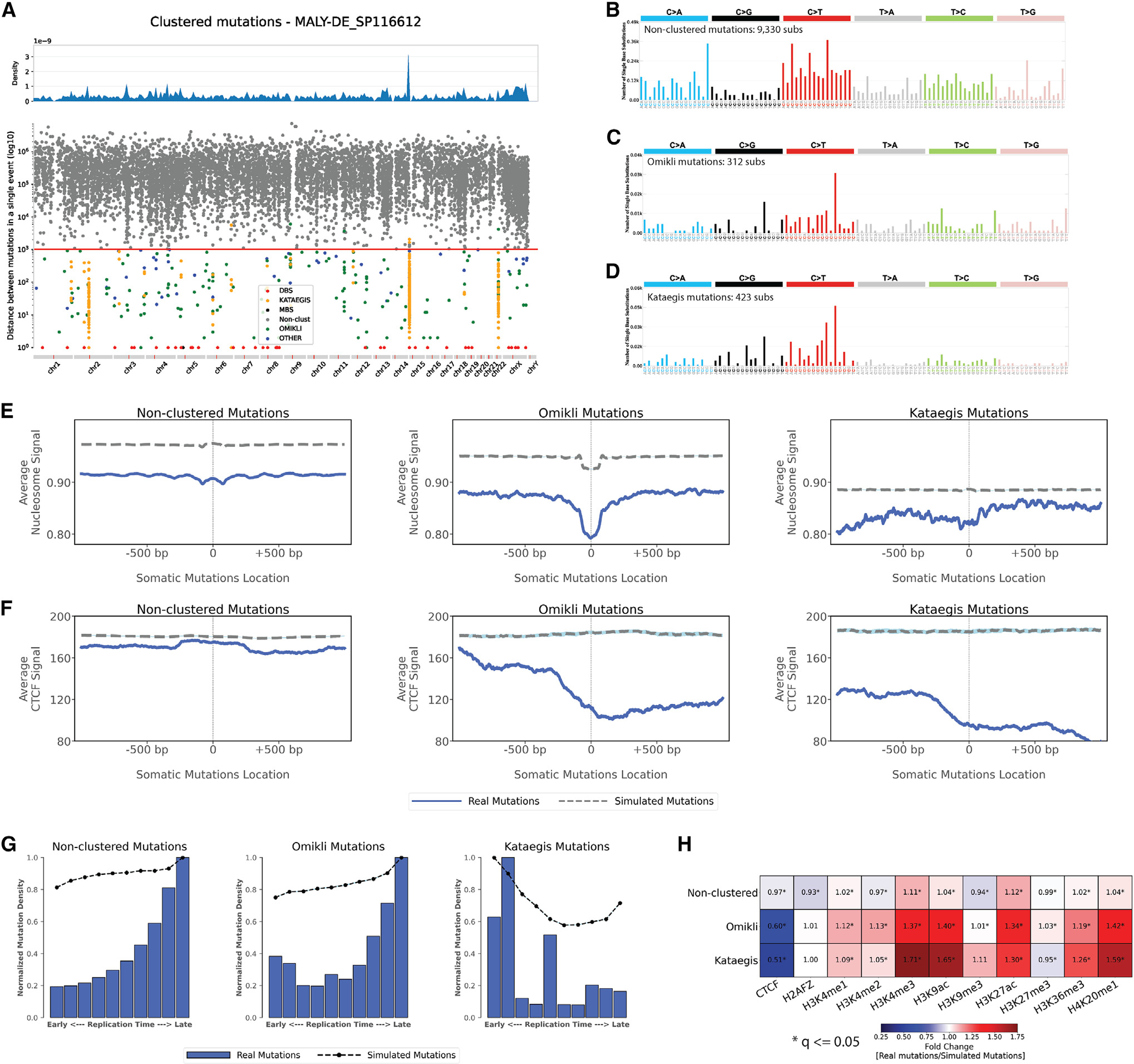
The somatic mutations found in a cancer genome are imprinted by different mutational processes. Each process exhibits a characteristic mutational signature, which can be affected by the genome architecture. However, the interplay between mutational signatures and topographical genomic features has not been extensively explored. Here, we integrate mutations from 5,120 whole-genome-sequenced tumors from 40 cancer types with 516 topographical features from ENCODE to evaluate the effect of nucleosome occupancy, histone modifications, CTCF binding, replication timing, and transcription/replication strand asymmetries on the cancer-specific accumulation of mutations from distinct mutagenic processes. Most mutational signatures are affected by topographical features, with signatures of related etiologies being similarly affected. Certain signatures exhibit periodic behaviors or cancer-type-specific enrichments/depletions near topographical features, revealing further information about the processes that imprinted them. Our findings, disseminated via the COSMIC (Catalog of Somatic Mutations in Cancer) signatures database, provide a comprehensive online resource for exploring the interactions between mutational signatures and topographical features across human cancer.
Keywords: CP: Cancer; CP: Genomics; cancer genomics; genome topography; mutational signatures; somatic mutations.
Copyright © 2023 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests L.B.A. is a compensated consultant and has equity interest in io9, LLC, and Genome Insight. His spouse is an employee of Biotheranostics, Inc. L.B.A. is also an inventor of US patent 10,776,718 for source identification by non-negative matrix factorization. E.N.B. and L.B.A. declare US provisional applications with serial numbers 63/289,601, 63/269,033, and 63/483,237. L.B.A. also declares US provisional applications with serial numbers 63/366,392, 63/412,835, and 63/492,348.
Figures

References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
