

Genome sequencing and comprehensive rare-variant analysis of 465 families with neurodevelopmental disorders
- PMID: 37541188
- PMCID: PMC10432178
- DOI: 10.1016/j.ajhg.2023.07.007
Genome sequencing and comprehensive rare-variant analysis of 465 families with neurodevelopmental disorders
Abstract
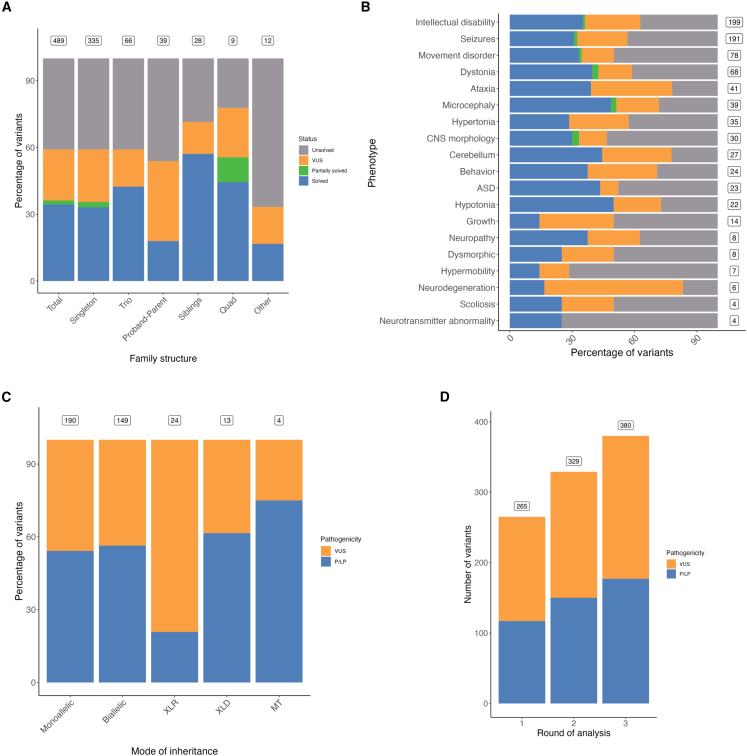
Despite significant progress in unraveling the genetic causes of neurodevelopmental disorders (NDDs), a substantial proportion of individuals with NDDs remain without a genetic diagnosis after microarray and/or exome sequencing. Here, we aimed to assess the power of short-read genome sequencing (GS), complemented with long-read GS, to identify causal variants in participants with NDD from the National Institute for Health and Care Research (NIHR) BioResource project. Short-read GS was conducted on 692 individuals (489 affected and 203 unaffected relatives) from 465 families. Additionally, long-read GS was performed on five affected individuals who had structural variants (SVs) in technically challenging regions, had complex SVs, or required distal variant phasing. Causal variants were identified in 36% of affected individuals (177/489), and a further 23% (112/489) had a variant of uncertain significance after multiple rounds of re-analysis. Among all reported variants, 88% (333/380) were coding nuclear SNVs or insertions and deletions (indels), and the remainder were SVs, non-coding variants, and mitochondrial variants. Furthermore, long-read GS facilitated the resolution of challenging SVs and invalidated variants of difficult interpretation from short-read GS. This study demonstrates the value of short-read GS, complemented with long-read GS, in investigating the genetic causes of NDDs. GS provides a comprehensive and unbiased method of identifying all types of variants throughout the nuclear and mitochondrial genomes in individuals with NDD.
Keywords: long-read sequencing; neurodevelopmental disorders; structural variants; whole-genome sequencing.
Copyright © 2023 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests K.J.C. and K.M. are currently employees of AstraZeneca.
Figures

References
-
- 100000 Genomes Project Pilot Investigators. Smedley D., Smith K.R., Martin A., Thomas E.A., McDonagh E.M., Cipriani V., Ellingford J.M., Arno G., Tucci A., et al. 100,000 Genomes pilot on rare-disease diagnosis in health care - preliminary report. N. Engl. J. Med. 2021;385:1868–1880. doi: 10.1056/NEJMoa2035790. - DOI - PMC - PubMed
-
- Clark M.M., Stark Z., Farnaes L., Tan T.Y., White S.M., Dimmock D., Kingsmore S.F. Meta-analysis of the diagnostic and clinical utility of genome and exome sequencing and chromosomal microarray in children with suspected genetic diseases. NPJ Genom. Med. 2018;3:16. doi: 10.1038/s41525-018-0053-8. - DOI - PMC - PubMed
-
- Belyeu J.R., Brand H., Wang H., Zhao X., Pedersen B.S., Feusier J., Gupta M., Nicholas T.J., Brown J., Baird L., et al. De novo structural mutation rates and gamete-of-origin biases revealed through genome sequencing of 2,396 families. Am. J. Hum. Genet. 2021;108:597–607. doi: 10.1016/j.ajhg.2021.02.012. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
