

Mammalian SWI/SNF chromatin remodeling complexes promote tyrosine kinase inhibitor resistance in EGFR-mutant lung cancer
- PMID: 37541244
- PMCID: PMC10957226
- DOI: 10.1016/j.ccell.2023.07.005
Mammalian SWI/SNF chromatin remodeling complexes promote tyrosine kinase inhibitor resistance in EGFR-mutant lung cancer
Abstract
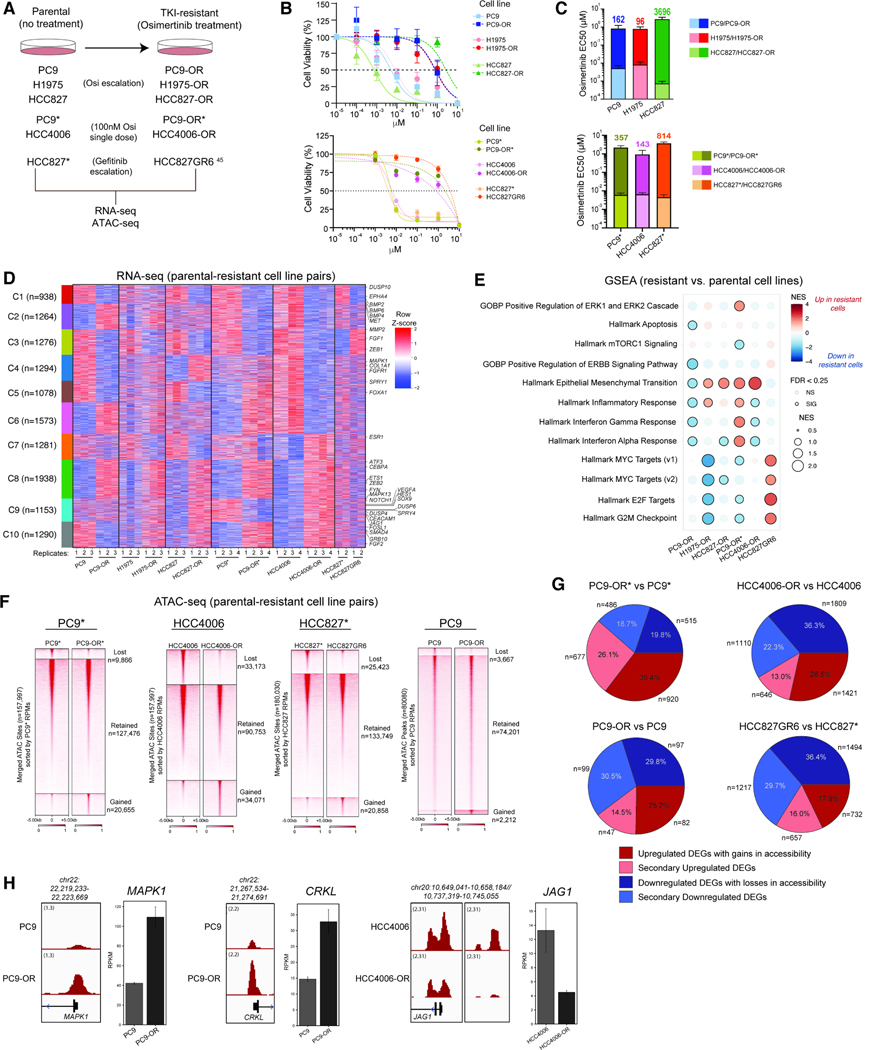
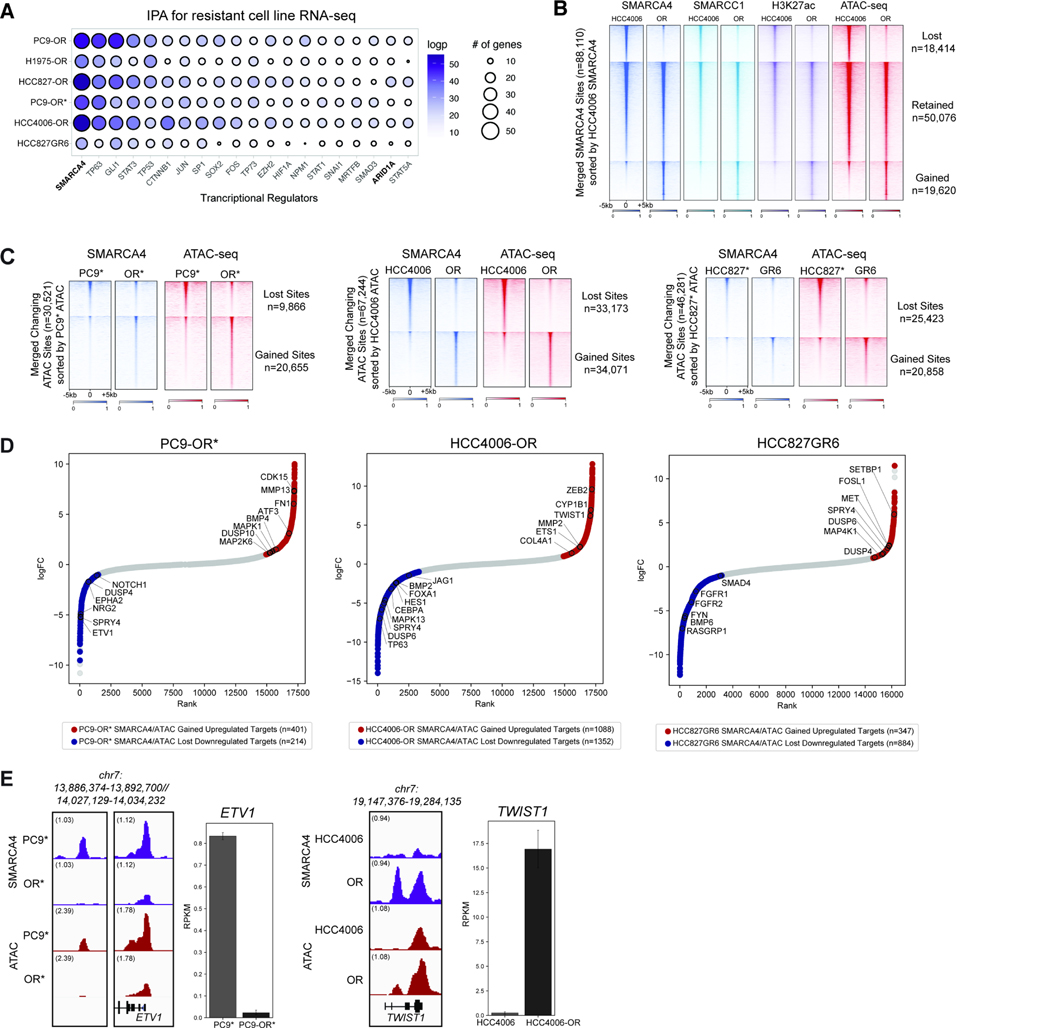
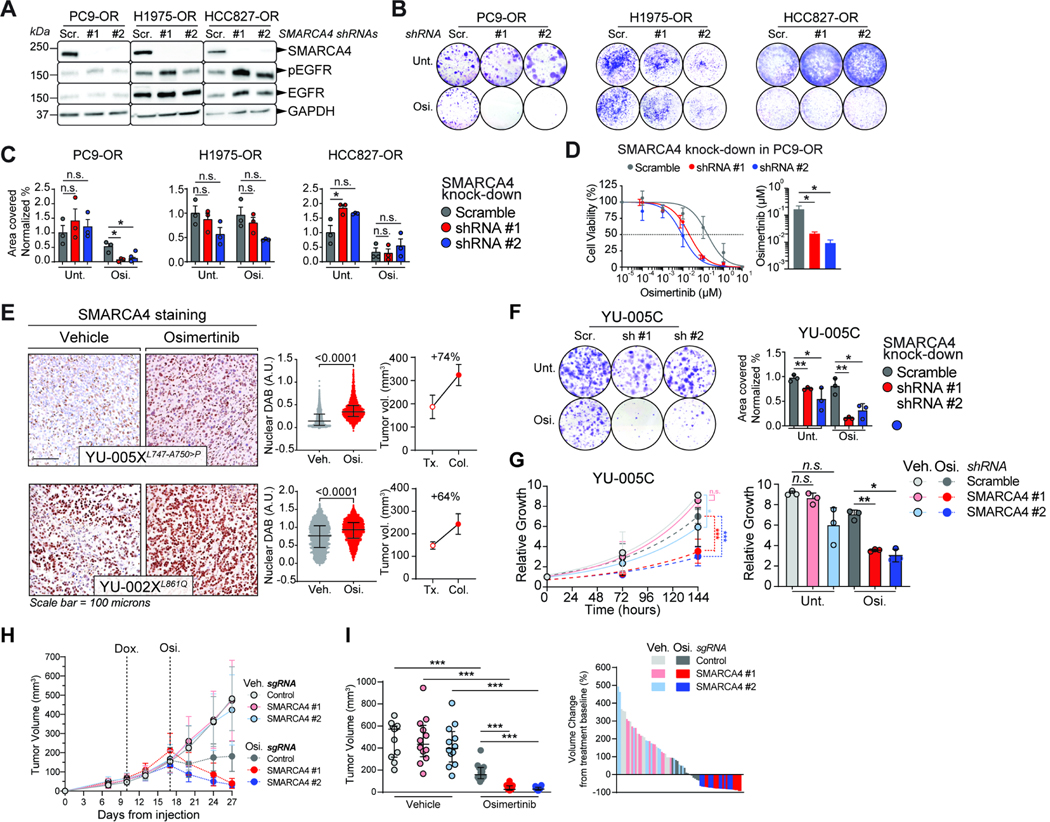
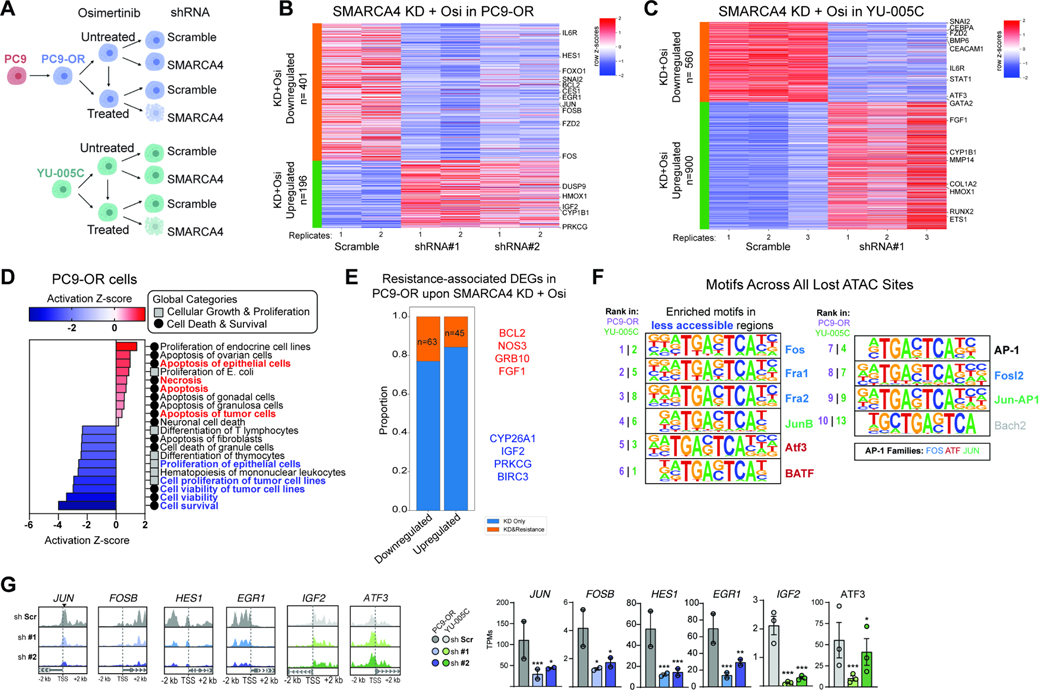
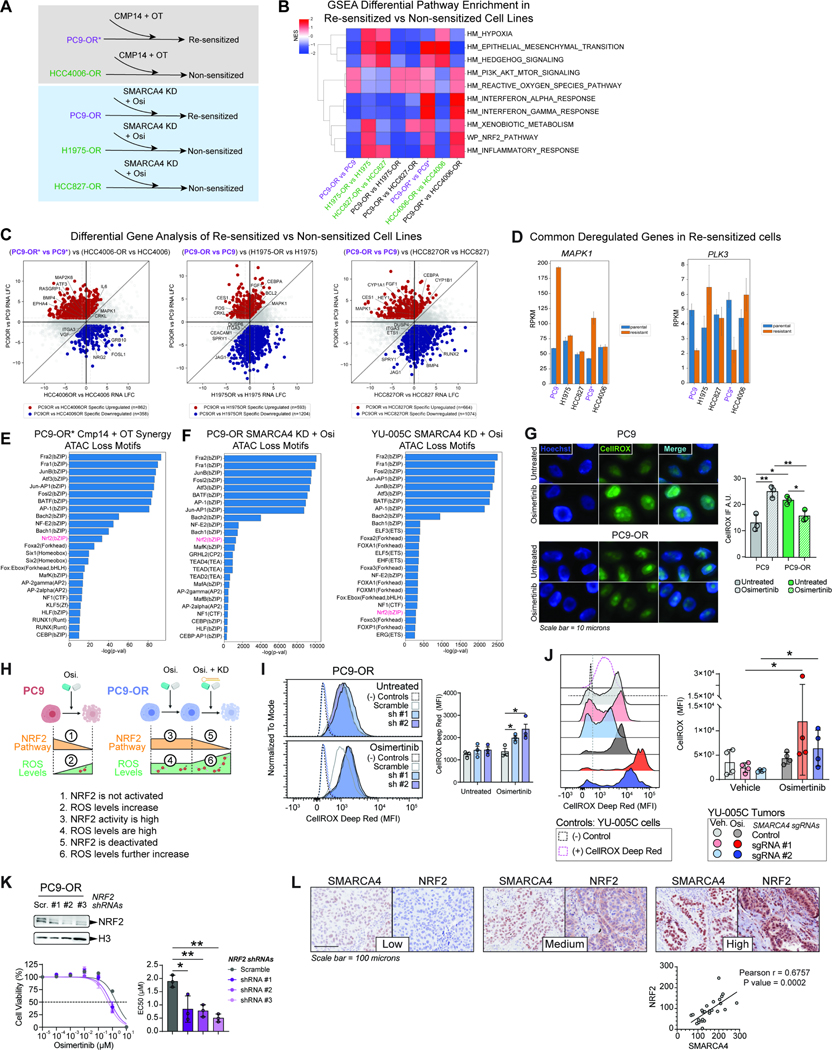
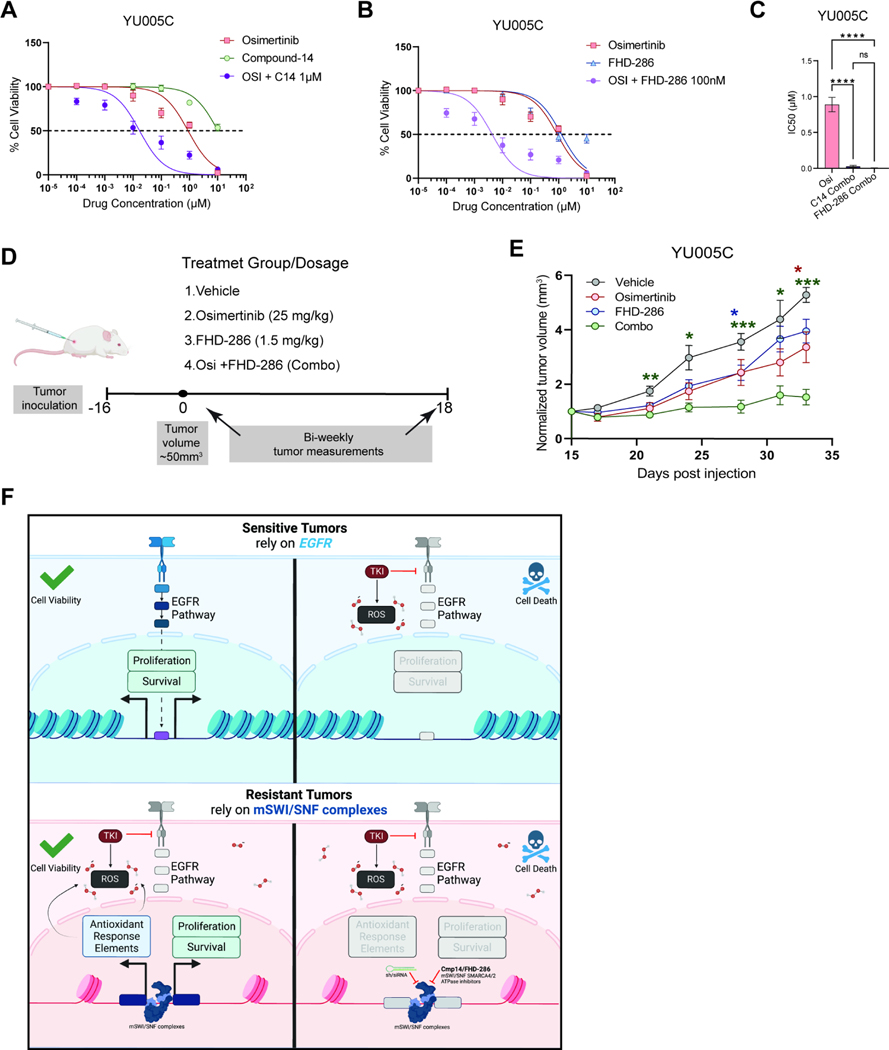
Acquired resistance to tyrosine kinase inhibitors (TKI), such as osimertinib used to treat EGFR-mutant lung adenocarcinomas, limits long-term efficacy and is frequently caused by non-genetic mechanisms. Here, we define the chromatin accessibility and gene regulatory signatures of osimertinib sensitive and resistant EGFR-mutant cell and patient-derived models and uncover a role for mammalian SWI/SNF chromatin remodeling complexes in TKI resistance. By profiling mSWI/SNF genome-wide localization, we identify both shared and cancer cell line-specific gene targets underlying the resistant state. Importantly, genetic and pharmacologic disruption of the SMARCA4/SMARCA2 mSWI/SNF ATPases re-sensitizes a subset of resistant models to osimertinib via inhibition of mSWI/SNF-mediated regulation of cellular programs governing cell proliferation, epithelial-to-mesenchymal transition, epithelial cell differentiation, and NRF2 signaling. These data highlight the role of mSWI/SNF complexes in supporting TKI resistance and suggest potential utility of mSWI/SNF inhibitors in TKI-resistant lung cancers.
Copyright © 2023 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests C.K. is the Scientific Founder, Scientific Advisor to the Board of Directors, Scientific Advisory Board member, shareholder, and consultant for Foghorn Therapeutics, Inc. (Cambridge, MA), serves on the Scientific Advisory Boards of Nereid Therapeutics, Nested Therapeutics, Accent Therapeutics, and Fibrogen, Inc. and is a consultant for Cell Signaling Technologies and Google Ventures. C.K. is also a member of the Molecular Cell and Cell Chemical Biology Editorial Boards. D.L. Rimm reports grants and personal fees from Amgen, Astra Zeneca, Cepheid, Konica – Minolta, Lilly, NextCure and personal fees from Cell Signaling Technology, Danaher, Fluidigm, GSK, Merck, Monopteros, NanoString, Odonate, Paige.AI, Regeneron, Roche, Sanofi, Ventana, Verily. K. Politi reports grants from the NCI/NIH; grants and personal fees from AstraZeneca; grants from Kolltan, Roche/Genentech, Boehringer Ingelheim, D2G Oncology and Symphogen; and personal fees from Janssen, Dynamo Therapeutics, Halda, Maverick Therapeutics, and Tocagen; and a patent for EGFR(T790M) mutation testing issued, licensed, and with royalties paid from Molecular Diagnostics/Memorial Sloan Kettering Cancer Center. P.A.J. is an equity owner in Gatekeeper Pharmaceuticals; consults for AstraZeneca, Boehringer Ingelheim, Pfizer, Roche/Genentech, Chugai Pharmaceuticals, Eli Lilly Pharmaceuticals, Araxes Pharmaceuticals, SFJ Pharmaceuticals, Voronoi, Daiichi Sankyo, Biocartis, Novartis, Sanofi, Takeda Oncology, Mirati Therapeutics, Transcenta, Silicon Therapeutics, Syndax, Nuvalent, Bayer, Esai, Allorion Therapeutics, Accutar Biotech, and Abbvie; receives research support from AstraZeneca, Daiichi Sankyo, PUMA, Eli Lilly, Boehringer Ingelheim, Revolution Medicines, and Takeda Oncology and is a co-inventor and receives postmarketing royalties on a DFCI owned patent on EGFR mutations licensed to LabCorp. Q.Y. reports grants and personal fees from AstraZeneca, and is a Scientific Advisory Board member of AccuraGen Inc. D.X.N received research funding from AstraZeneca. The other authors declare no competing interests.
Figures

References
-
- Kim G, McKee AE, Ning YM, Hazarika M, Theoret M, Johnson JR, Xu QC, Tang S, Sridhara R, Jiang X, et al. (2014). FDA approval summary: vemurafenib for treatment of unresectable or metastatic melanoma with the BRAFV600E mutation. Clin Cancer Res 20, 4994–5000. 10.1158/1078-0432.CCR-14-0776. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
