

Targeting of SLC25A22 boosts the immunotherapeutic response in KRAS-mutant colorectal cancer
- PMID: 37542037
- PMCID: PMC10403583
- DOI: 10.1038/s41467-023-39571-6
Targeting of SLC25A22 boosts the immunotherapeutic response in KRAS-mutant colorectal cancer
Abstract
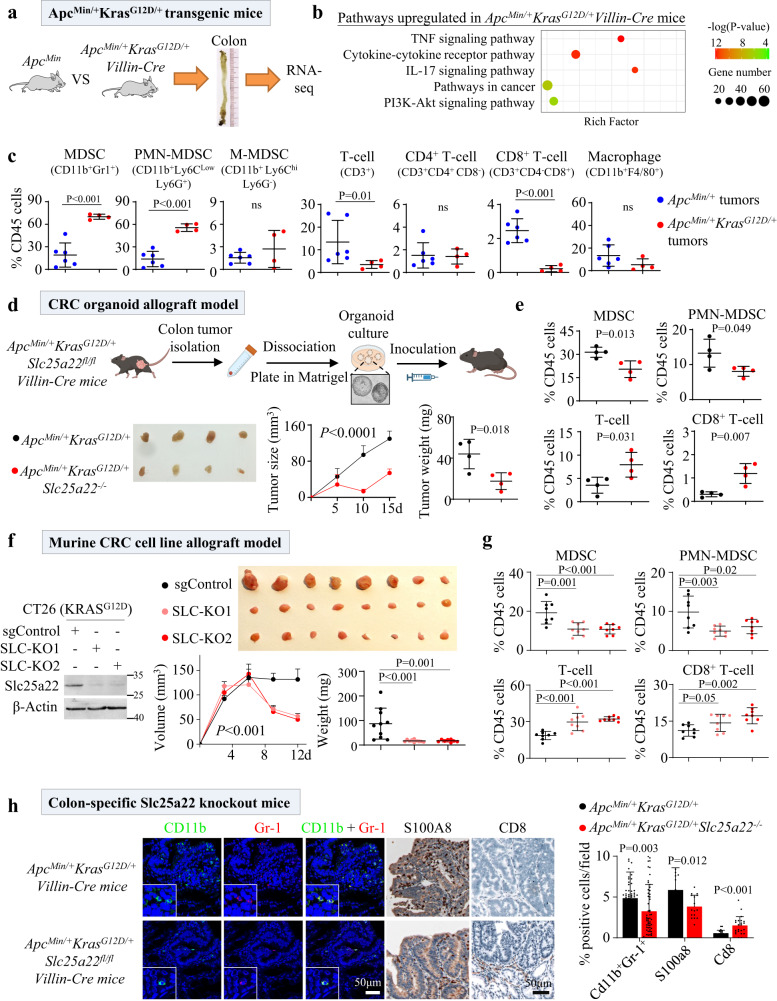
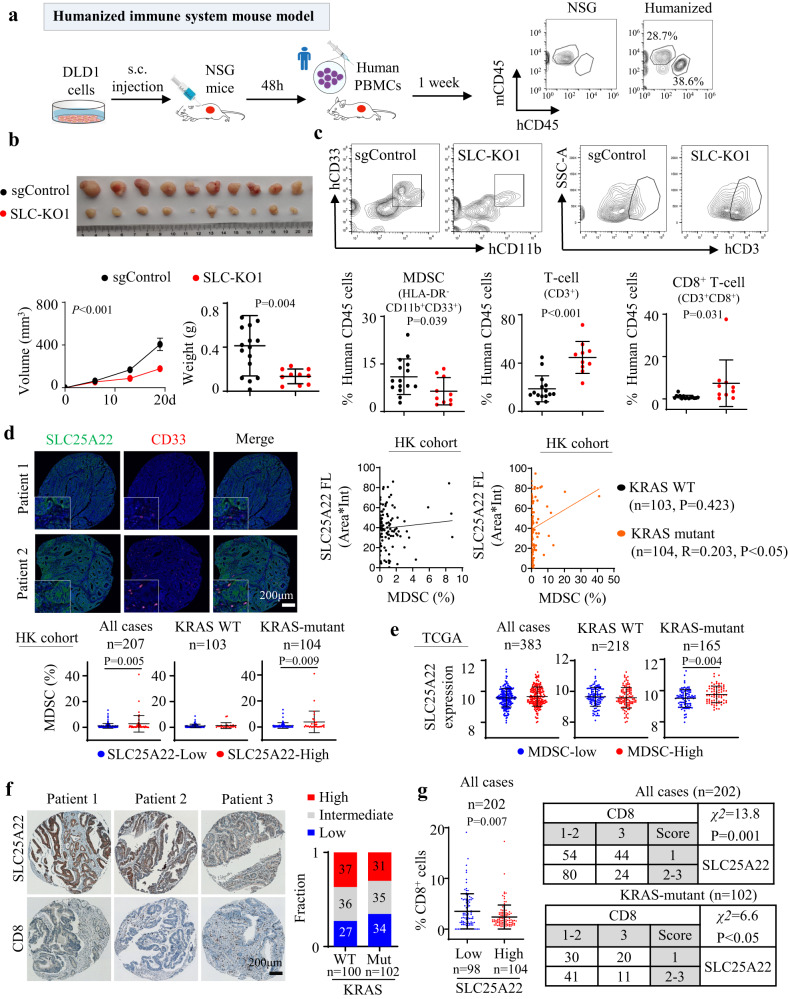
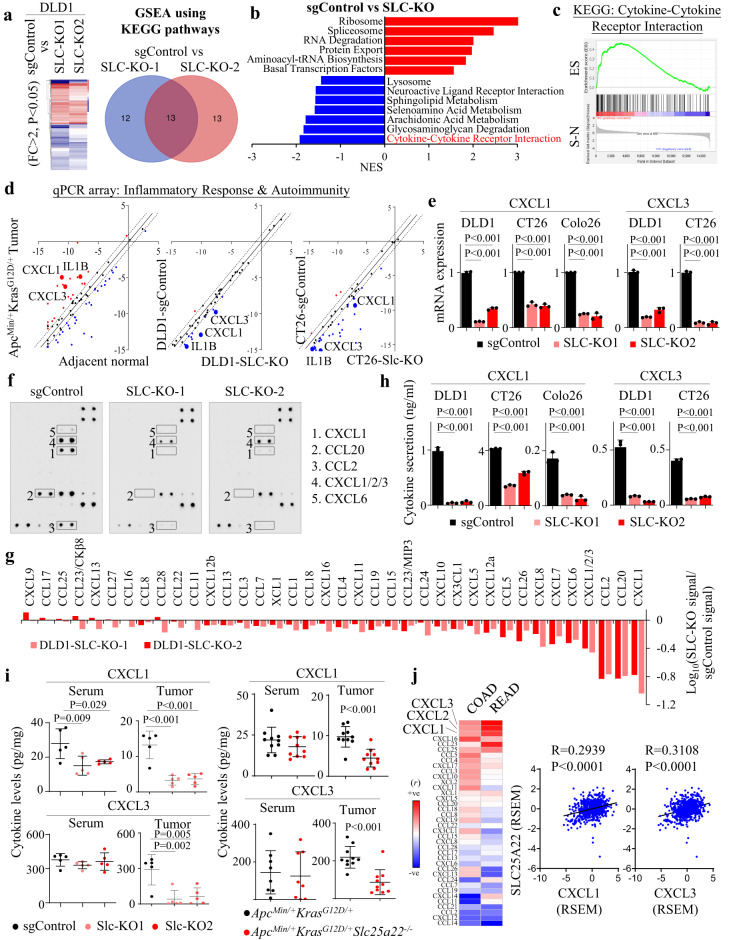
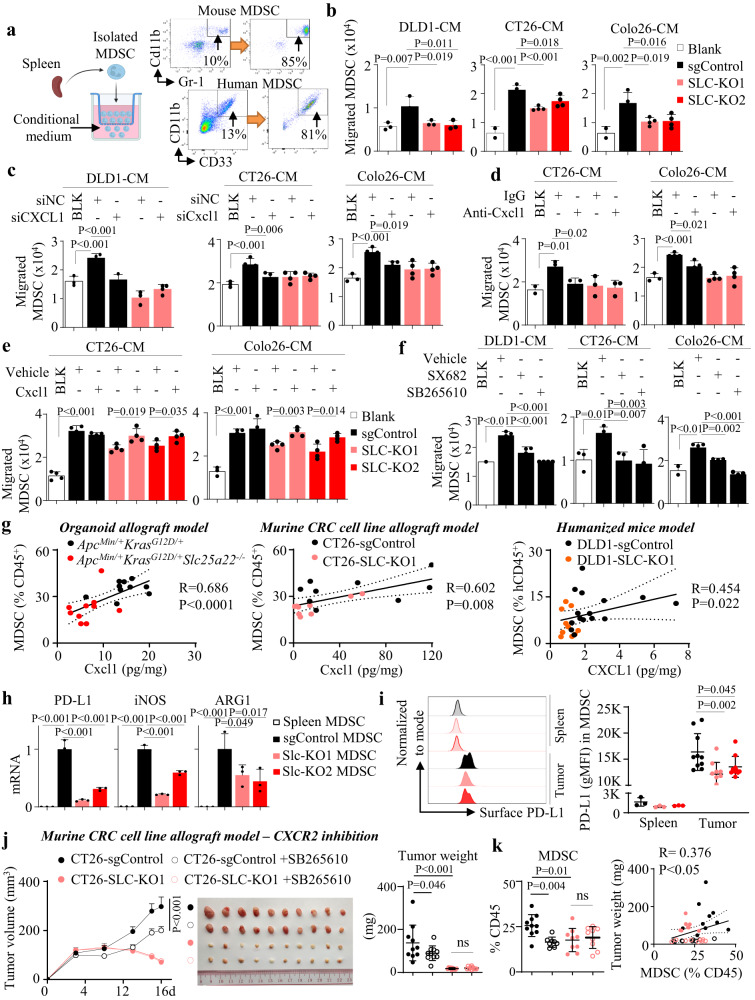
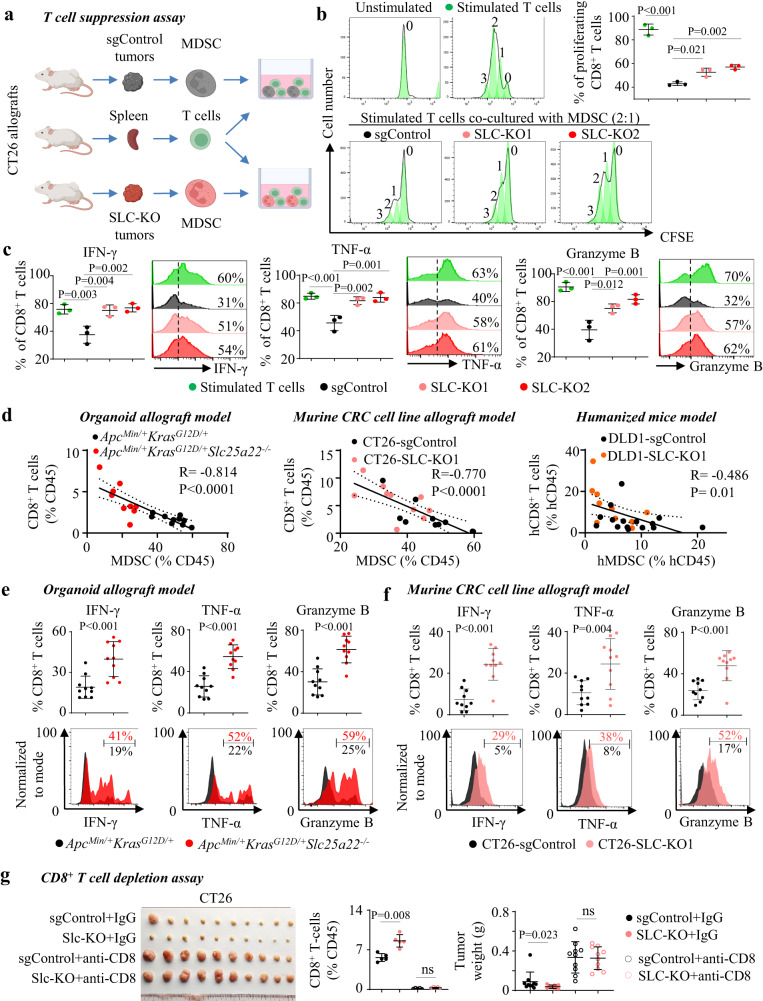
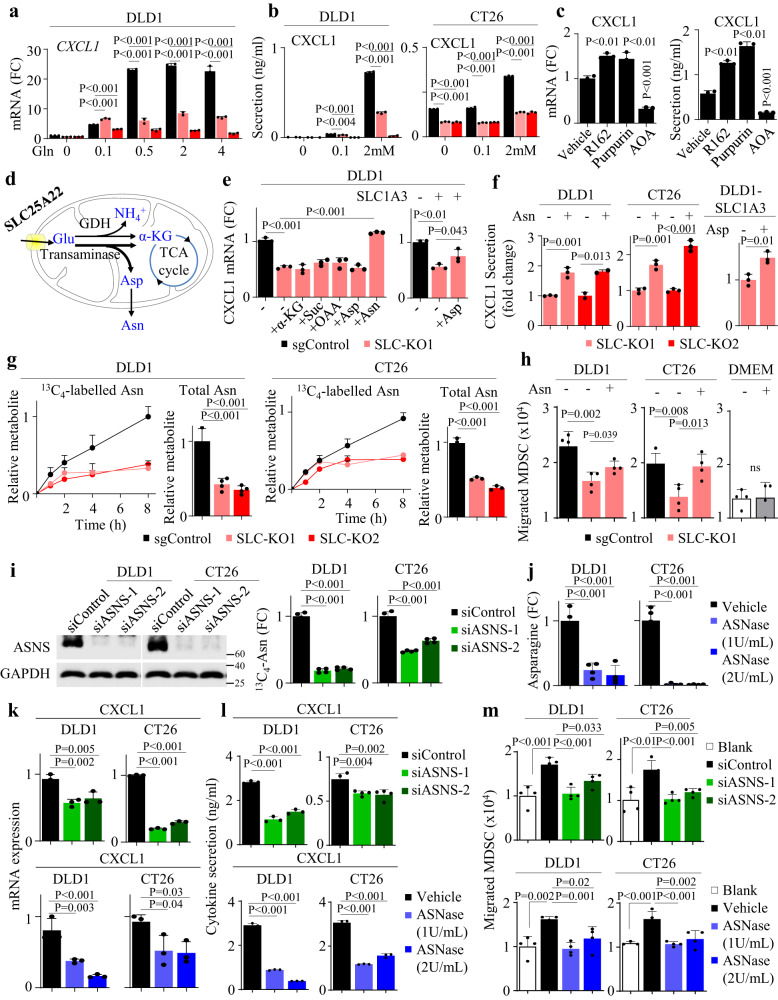
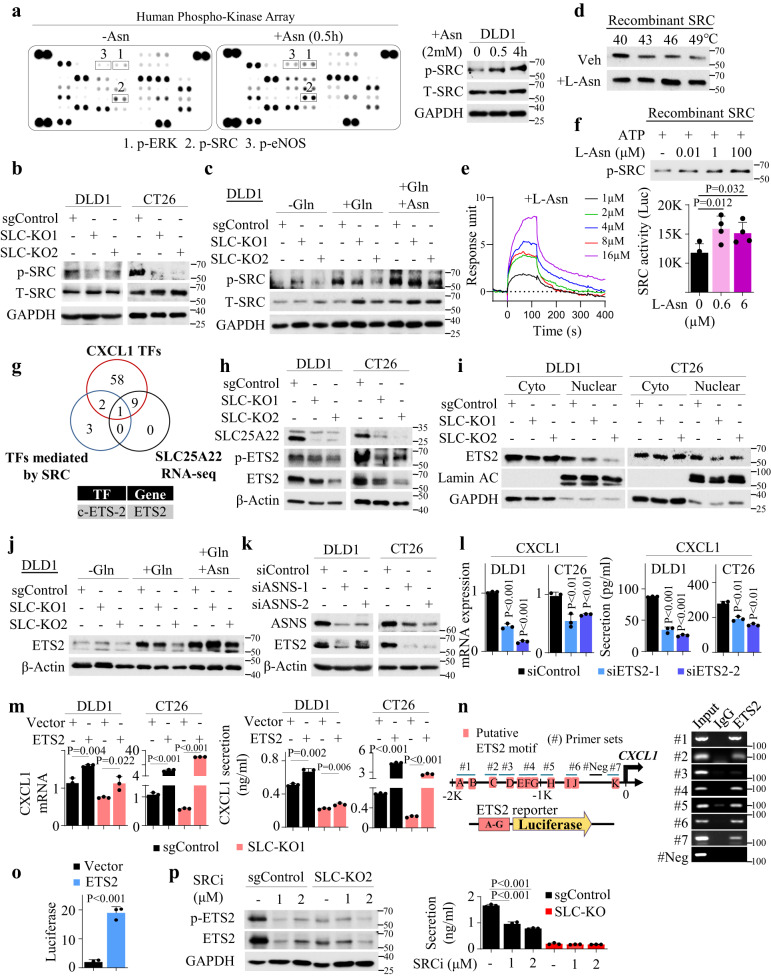
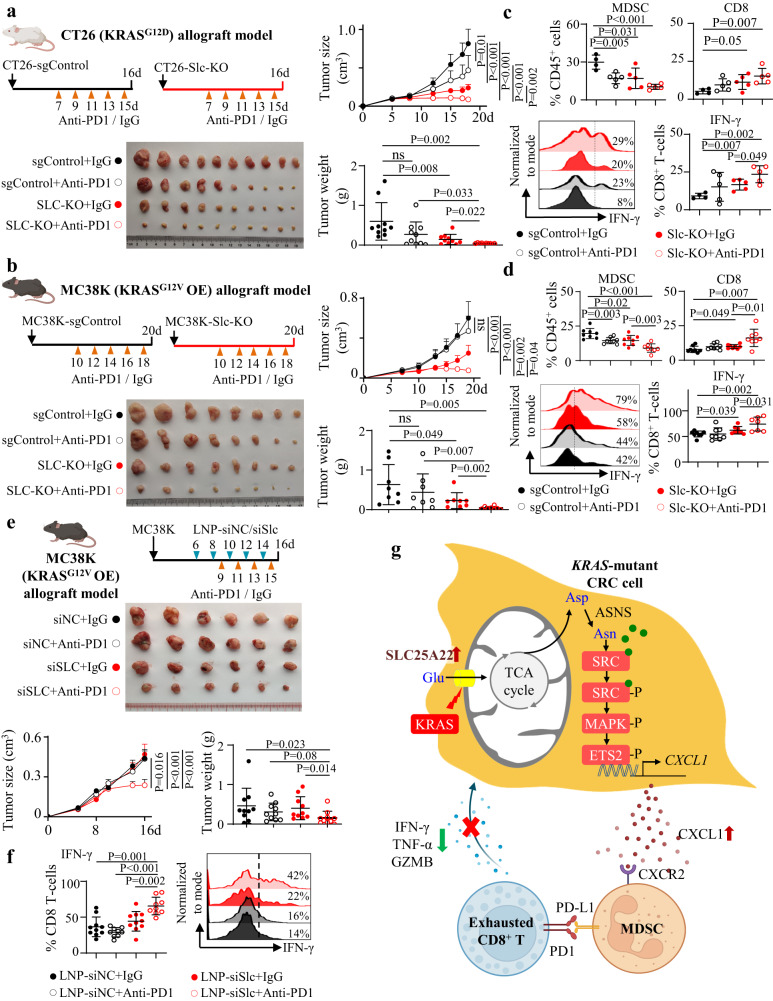
KRAS is an important tumor intrinsic factor driving immune suppression in colorectal cancer (CRC). In this study, we demonstrate that SLC25A22 underlies mutant KRAS-induced immune suppression in CRC. In immunocompetent male mice and humanized male mice models, SLC25A22 knockout inhibits KRAS-mutant CRC tumor growth with reduced myeloid derived suppressor cells (MDSC) but increased CD8+ T-cells, implying the reversion of mutant KRAS-driven immunosuppression. Mechanistically, we find that SLC25A22 plays a central role in promoting asparagine, which binds and activates SRC phosphorylation. Asparagine-mediated SRC promotes ERK/ETS2 signaling, which drives CXCL1 transcription. Secreted CXCL1 functions as a chemoattractant for MDSC via CXCR2, leading to an immunosuppressive microenvironment. Targeting SLC25A22 or asparagine impairs KRAS-induced MDSC infiltration in CRC. Finally, we demonstrate that the targeting of SLC25A22 in combination with anti-PD1 therapy synergizes to inhibit MDSC and activate CD8+ T cells to suppress KRAS-mutant CRC growth in vivo. We thus identify a metabolic pathway that drives immunosuppression in KRAS-mutant CRC.
© 2023. Springer Nature Limited.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
