

Immune aging - A mechanism in autoimmune disease
- PMID: 37542986
- PMCID: PMC10663095
- DOI: 10.1016/j.smim.2023.101814
Immune aging - A mechanism in autoimmune disease
Abstract
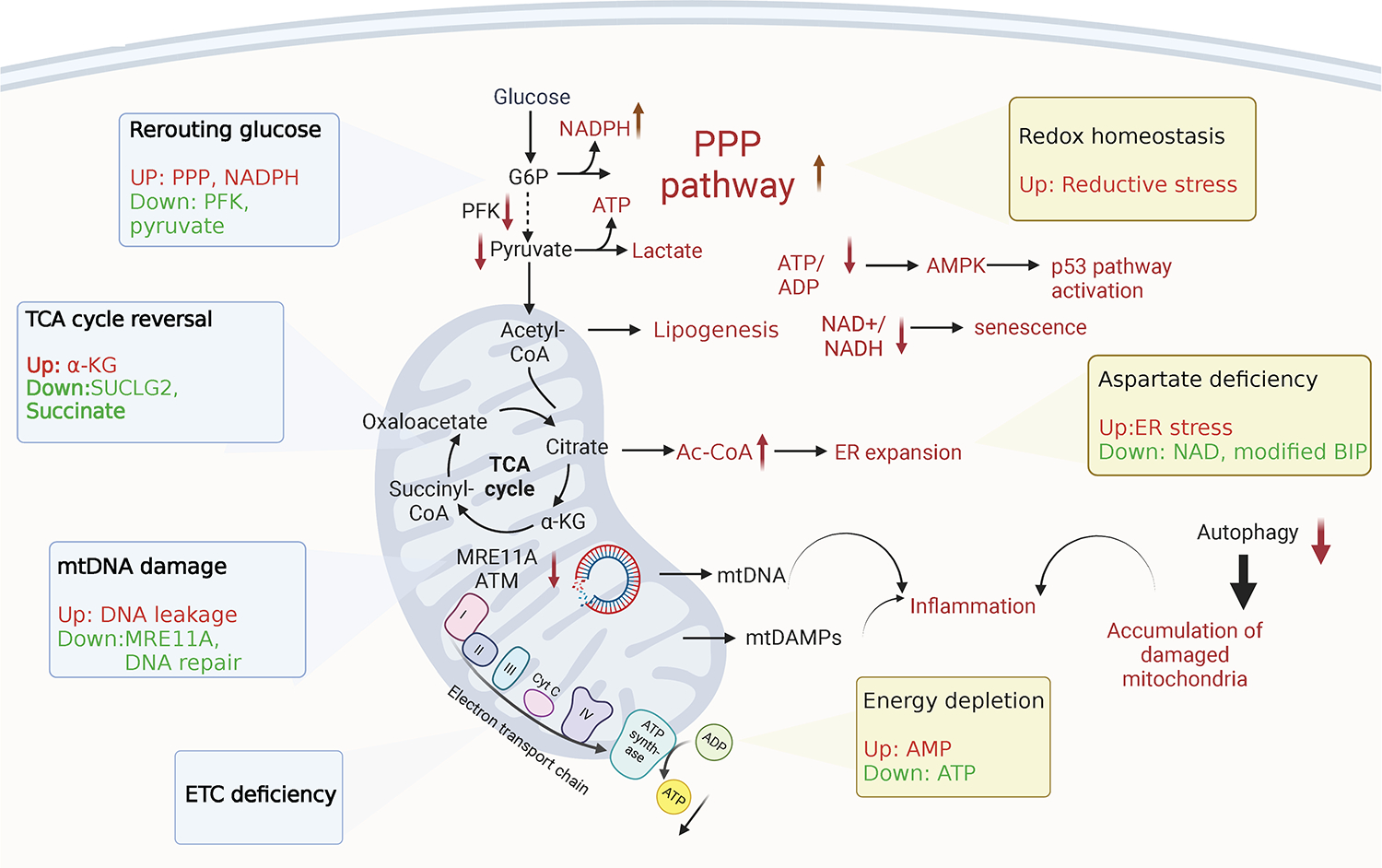
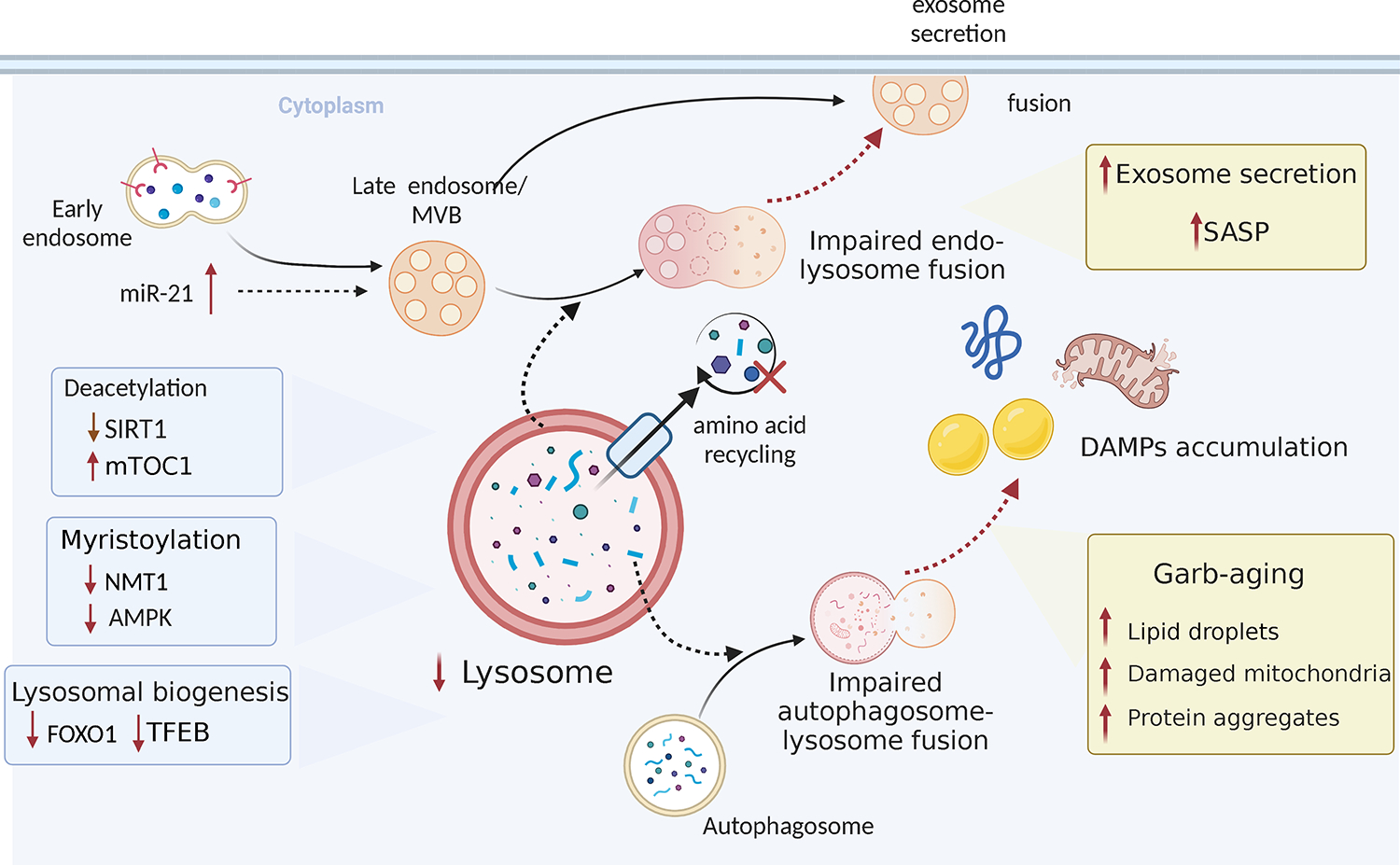
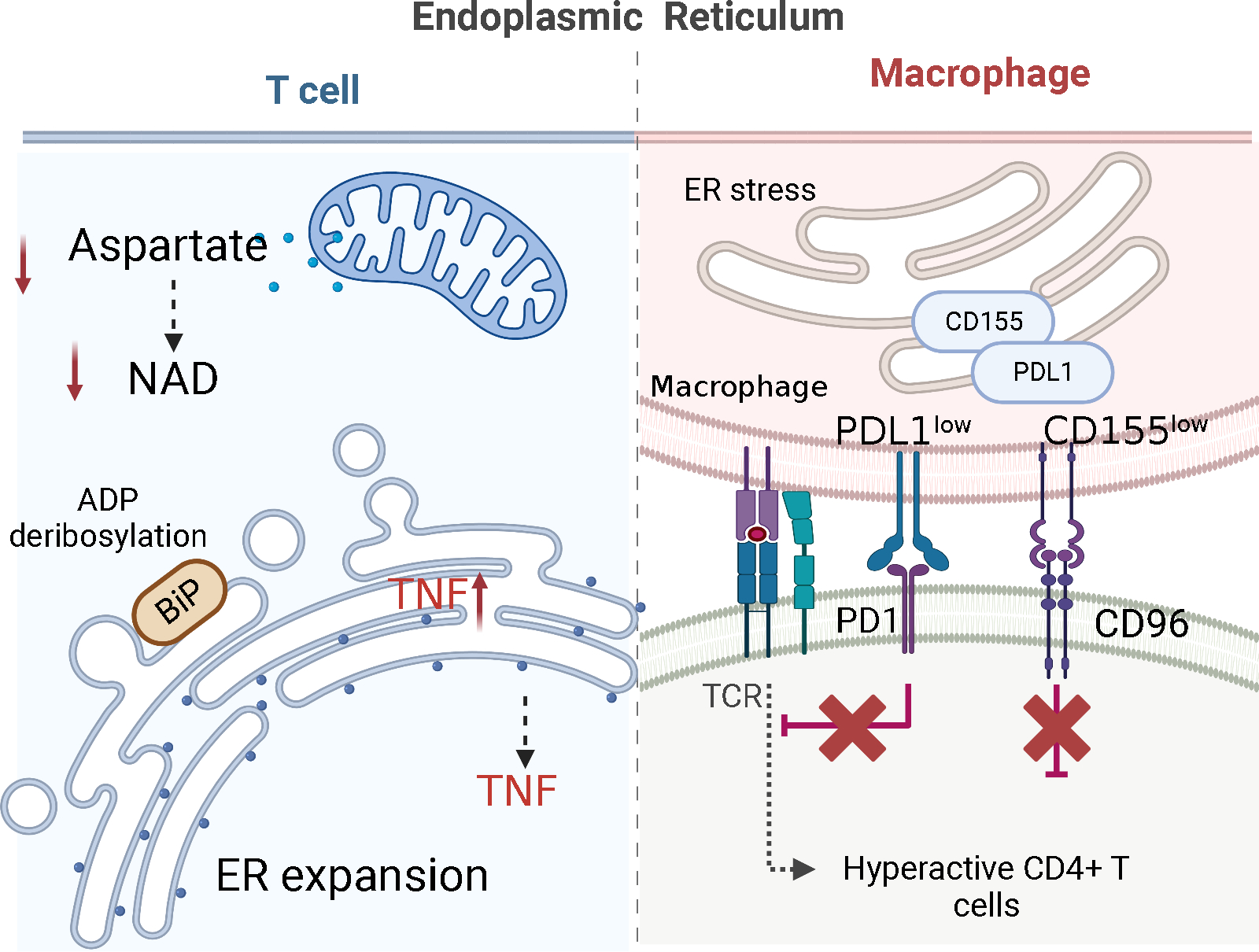
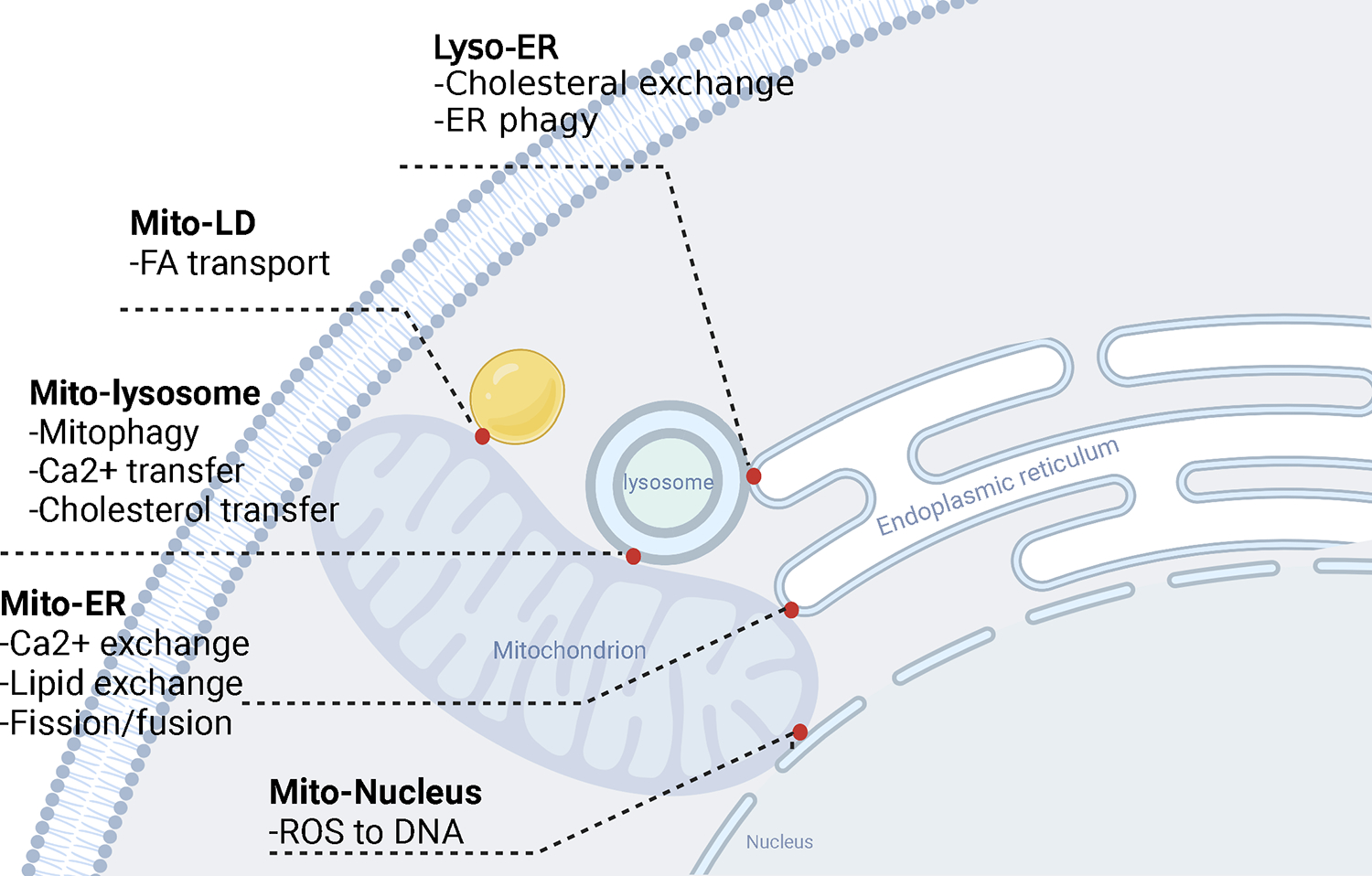
Evidence is emerging that the process of immune aging is a mechanism leading to autoimmunity. Over lifetime, the immune system adapts to profound changes in hematopoiesis and lymphogenesis, and progressively restructures in face of an ever-expanding exposome. Older adults fail to generate adequate immune responses against microbial infections and tumors, but accumulate aged T cells, B cells and myeloid cells. Age-associated B cells are highly efficient in autoantibody production. T-cell aging promotes the accrual of end-differentiated effector T cells with potent cytotoxic and pro-inflammatory abilities and myeloid cell aging supports a low grade, sterile and chronic inflammatory state (inflammaging). In pre-disposed individuals, immune aging can lead to frank autoimmune disease, manifesting with chronic inflammation and irreversible tissue damage. Emerging data support the concept that autoimmunity results from aging-induced failure of fundamental cellular processes in immune effector cells: genomic instability, loss of mitochondrial fitness, failing proteostasis, dwindling lysosomal degradation and inefficient autophagy. Here, we have reviewed the evidence that malfunctional mitochondria, disabled lysosomes and stressed endoplasmic reticula induce pathogenic T cells and macrophages that drive two autoimmune diseases, rheumatoid arthritis (RA) and giant cell arteritis (GCA). Recognizing immune aging as a risk factor for autoimmunity will open new avenues of immunomodulatory therapy, including the repair of malfunctioning mitochondria and lysosomes.
Keywords: Autoimmune disease; Giant cell arteritis; Immune aging; Rheumatoid arthritis; T cell aging.
Copyright © 2023. Published by Elsevier Ltd.
Conflict of interest statement
Declaration of Competing Interest The authors declare no conflicts of interest.
Figures
References
-
- Berner F, Bomze D, Lichtensteiger C, Walter V, Niederer R, Hasan Ali O et al. Autoreactive napsin A-specific T cells are enriched in lung tumors and inflammatory lung lesions during immune checkpoint blockade. Sci Immunol, 2022;7:eabn9644. - PubMed
-
- Cancro MP. Age-Associated B Cells. Annu Rev Immunol, 2020;38:315–40. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
