

Multi-omics profiling of CSF from spinal muscular atrophy type 3 patients after nusinersen treatment: a 2-year follow-up multicenter retrospective study
- PMID: 37543540
- PMCID: PMC10404194
- DOI: 10.1007/s00018-023-04885-7
Multi-omics profiling of CSF from spinal muscular atrophy type 3 patients after nusinersen treatment: a 2-year follow-up multicenter retrospective study
Abstract
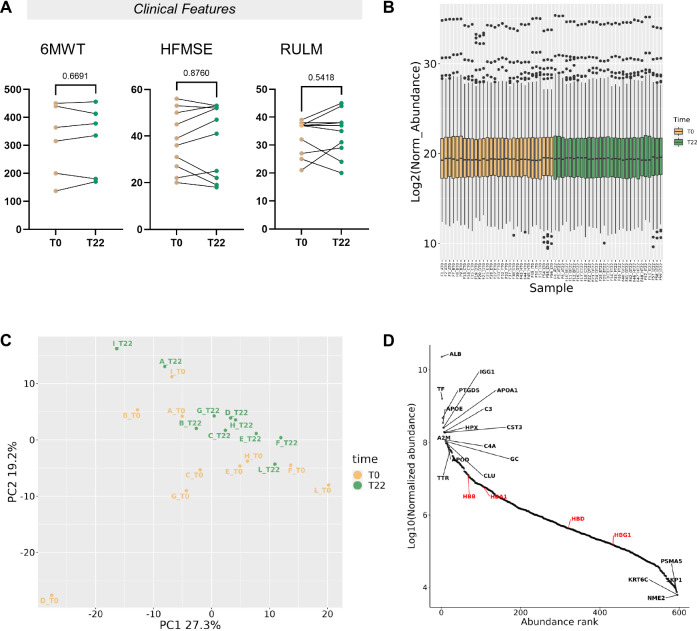
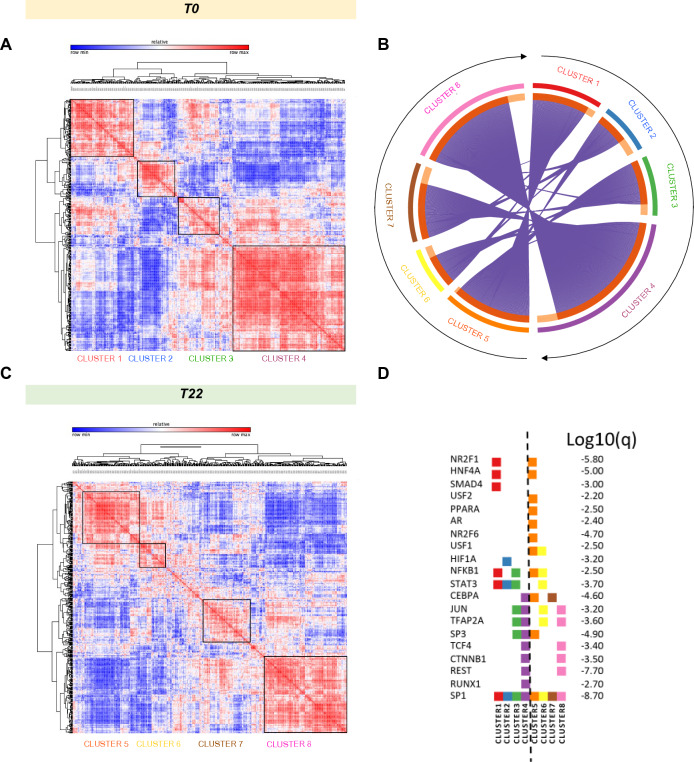
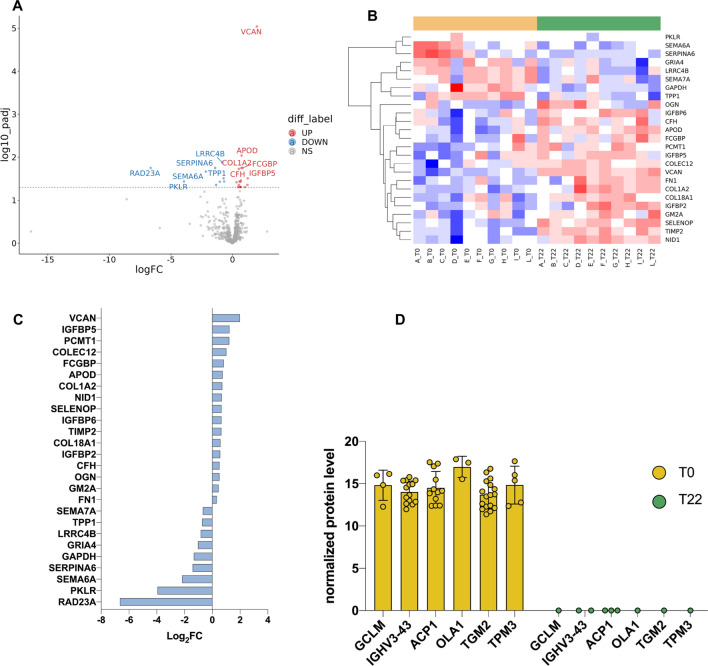
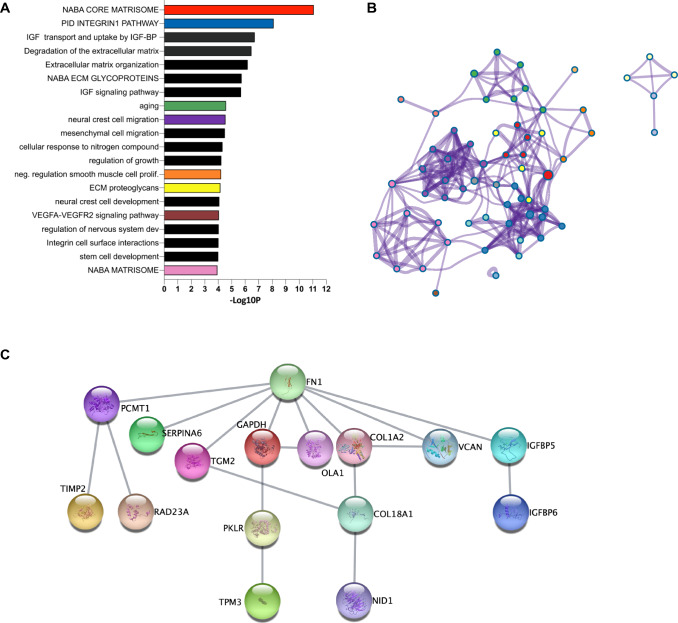
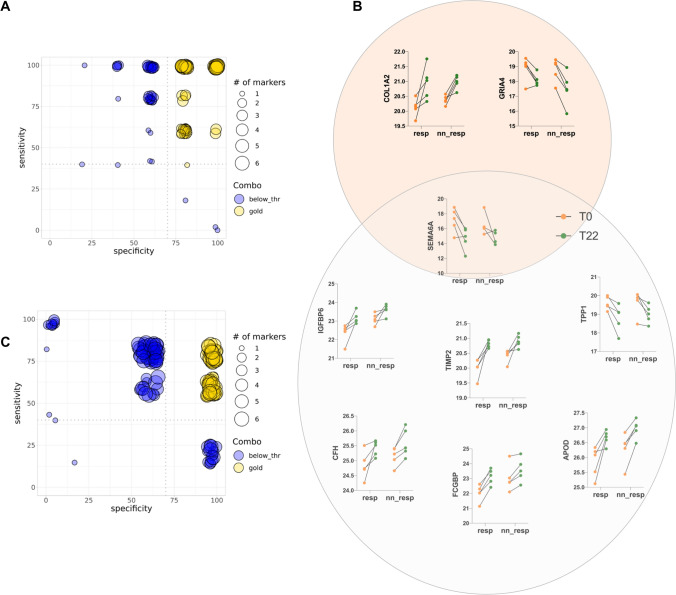
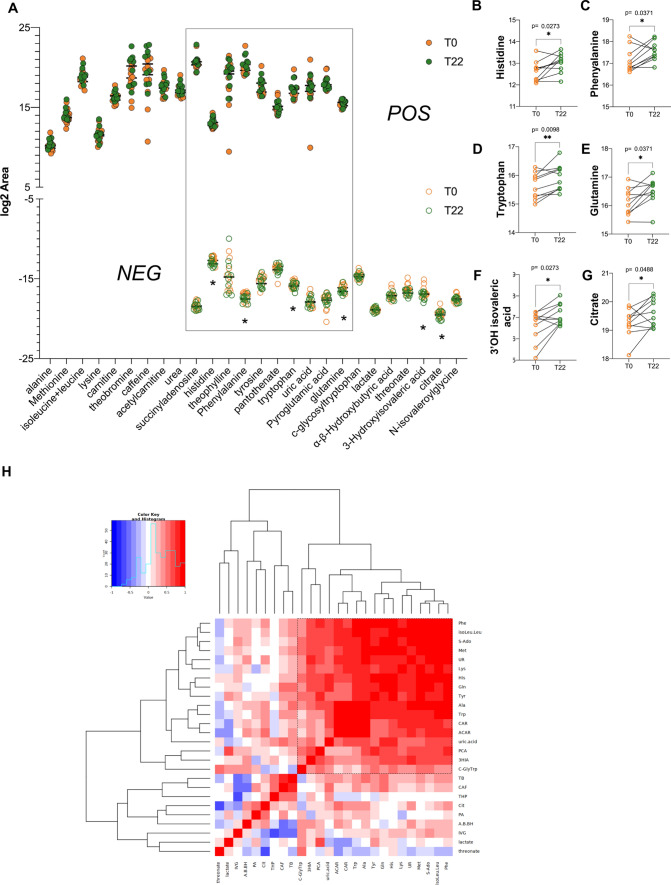
Spinal muscular atrophy (SMA) is a neurodegenerative disorder caused by mutations in the SMN1 gene resulting in reduced levels of the SMN protein. Nusinersen, the first antisense oligonucleotide (ASO) approved for SMA treatment, binds to the SMN2 gene, paralogue to SMN1, and mediates the translation of a functional SMN protein. Here, we used longitudinal high-resolution mass spectrometry (MS) to assess both global proteome and metabolome in cerebrospinal fluid (CSF) from ten SMA type 3 patients, with the aim of identifying novel readouts of pharmacodynamic/response to treatment and predictive markers of treatment response. Patients had a median age of 33.5 [29.5; 38.25] years, and 80% of them were ambulant at time of the enrolment, with a median HFMSE score of 37.5 [25.75; 50.75]. Untargeted CSF proteome and metabolome were measured using high-resolution MS (nLC-HRMS) on CSF samples obtained before treatment (T0) and after 2 years of follow-up (T22). A total of 26 proteins were found to be differentially expressed between T0 and T22 upon VSN normalization and LIMMA differential analysis, accounting for paired replica. Notably, key markers of the insulin-growth factor signaling pathway were upregulated after treatment together with selective modulation of key transcription regulators. Using CombiROC multimarker signature analysis, we suggest that detecting a reduction of SEMA6A and an increase of COL1A2 and GRIA4 might reflect therapeutic efficacy of nusinersen. Longitudinal metabolome profiling, analyzed with paired t-Test, showed a significant shift for some aminoacid utilization induced by treatment, whereas other metabolites were largely unchanged. Together, these data suggest perturbation upon nusinersen treatment still sustained after 22 months of follow-up and confirm the utility of CSF multi-omic profiling as pharmacodynamic biomarker for SMA type 3. Nonetheless, validation studies are needed to confirm this evidence in a larger sample size and to further dissect combined markers of response to treatment.
Keywords: Antisense oligonucleotides; Metabolomic; Proteomic; Spinal muscular atrophy.
© 2023. The Author(s).
Conflict of interest statement
The authors confirm that there are no conflicts of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
