

Mass spectrometry-based proteomic characterization of the middle-aged mouse brain for animal model research of neuromuscular diseases
- PMID: 37545360
- PMCID: PMC10583138
- DOI: 10.4081/ejtm.2023.11553
Mass spectrometry-based proteomic characterization of the middle-aged mouse brain for animal model research of neuromuscular diseases
Abstract
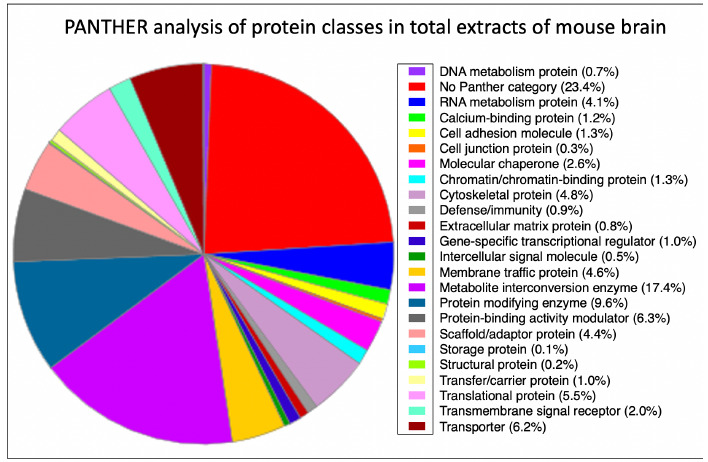
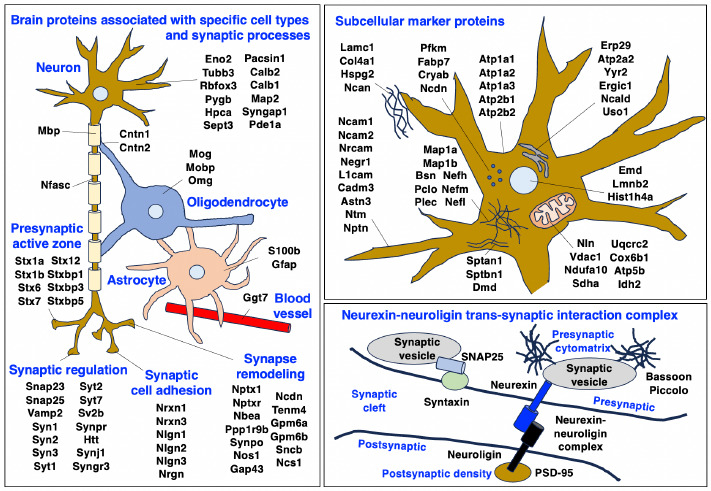
Neuromuscular diseases with primary muscle wasting symptoms may also display multi-systemic changes in the body and exhibit secondary pathophysiological alterations in various non-muscle tissues. In some cases, this includes proteome-wide alterations and/or adaptations in the central nervous system. Thus, in order to provide an improved bioanalytical basis for the comprehensive evaluation of animal models that are routinely used in muscle research, this report describes the mass spectrometry-based proteomic characterization of the mouse brain. Crude tissue extracts were examined by bottom-up proteomics and detected 4558 distinct protein species. The detailed analysis of the brain proteome revealed the presence of abundant cellular proteoforms in the neuronal cytoskeleton, as well as various brain region enriched proteins, including markers of the cerebral cortex, cerebellum, hippocampus and the olfactory bulb. Neuroproteomic markers of specific cell types in the brain were identified in association with various types of neurons and glia cells. Markers of subcellular structures were established for the plasmalemma, nucleus, endoplasmic reticulum, mitochondria and other crucial organelles, as well as synaptic components that are involved in presynaptic vesicle docking, neurotransmitter release and synapse remodelling.
Conflict of interest statement
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Another example of a neuromuscular disorder with complex body-wide alterations including abnormal brain functions is Duchenne muscular dystrophy (DMD). This X-linked inherited disorder can be clearly defined as a primary muscle wasting disorder but can also be categorized as a multi-systemic disease. Dystrophinopathies are due to mutations in the
In order to establish the scientific basis for comparative proteomic surveys of the central nervous system in relation to mouse model research of neuromuscular diseases, this report outlines the mass spectrometry-based proteomic characterization of the normal middle-aged mouse brain. The proteomic data of markers of brain regions such as the olfactory bulb, hippocampus, cerebellum, hypothalamus and cerebral cortex, various brain cell types and subcellular structures found in neurons, synapsis and glia cells should be helpful as a reference guide for the comprehensive evaluation of the central nervous system in genetic mouse models that are routinely used in basic and applied myology research.
Figures
References
-
- Brown RH, Al-Chalabi A. Amyotrophic Lateral Sclerosis. N Engl J Med. 2017. Jul 13;377(2):162-172. doi: 10.1056/NEJMra1603471. PMID: 28700839. - PubMed
LinkOut - more resources
Full Text Sources
