

This is a preprint.
Systematic assessment of long-read RNA-seq methods for transcript identification and quantification
- PMID: 37546854
- PMCID: PMC10402094
- DOI: 10.1101/2023.07.25.550582
Systematic assessment of long-read RNA-seq methods for transcript identification and quantification
Update in
-
Systematic assessment of long-read RNA-seq methods for transcript identification and quantification.Nat Methods. 2024 Jul;21(7):1349-1363. doi: 10.1038/s41592-024-02298-3. Epub 2024 Jun 7. Nat Methods. 2024. PMID: 38849569 Free PMC article.
Abstract
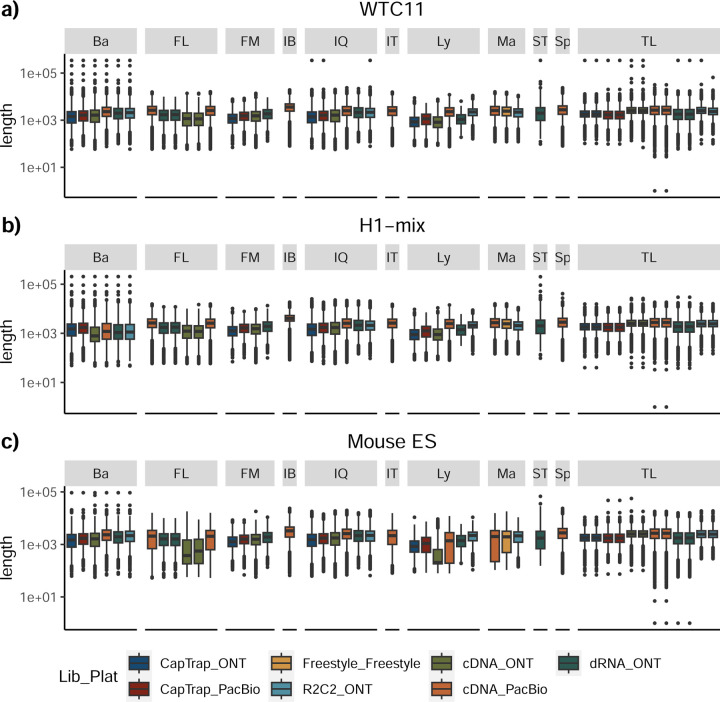
The Long-read RNA-Seq Genome Annotation Assessment Project (LRGASP) Consortium was formed to evaluate the effectiveness of long-read approaches for transcriptome analysis. The consortium generated over 427 million long-read sequences from cDNA and direct RNA datasets, encompassing human, mouse, and manatee species, using different protocols and sequencing platforms. These data were utilized by developers to address challenges in transcript isoform detection and quantification, as well as de novo transcript isoform identification. The study revealed that libraries with longer, more accurate sequences produce more accurate transcripts than those with increased read depth, whereas greater read depth improved quantification accuracy. In well-annotated genomes, tools based on reference sequences demonstrated the best performance. When aiming to detect rare and novel transcripts or when using reference-free approaches, incorporating additional orthogonal data and replicate samples are advised. This collaborative study offers a benchmark for current practices and provides direction for future method development in transcriptome analysis.
Conflict of interest statement
Competing Interests Design of the project was discussed with Oxford Nanopore Technologies (ONT), Pacific Biosciences, and Lexogen. ONT provided partial support for flow cells and reagents. S.C-S and A.N.B. have received reimbursement for travel, accommodation, and conference fees to speak at events organized by ONT. A.N.B. is a consultant for Remix Therapeutics, Inc.
Figures
















































































References
-
- Garalde D. R. et al. Highly parallel direct RNA sequencing on an array of nanopores. Nat. Methods 15, 201–206 (2018). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources