

This is a preprint.
ChromaFold predicts the 3D contact map from single-cell chromatin accessibility
- PMID: 37546906
- PMCID: PMC10402156
- DOI: 10.1101/2023.07.27.550836
ChromaFold predicts the 3D contact map from single-cell chromatin accessibility
Update in
-
ChromaFold predicts the 3D contact map from single-cell chromatin accessibility.Nat Commun. 2024 Nov 1;15(1):9432. doi: 10.1038/s41467-024-53628-0. Nat Commun. 2024. PMID: 39487131 Free PMC article.
Abstract
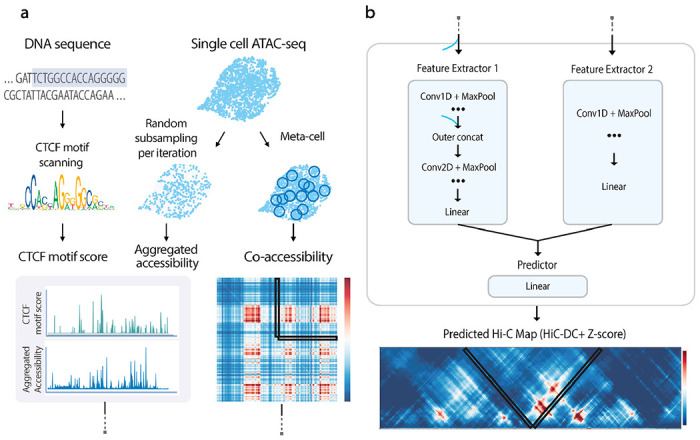
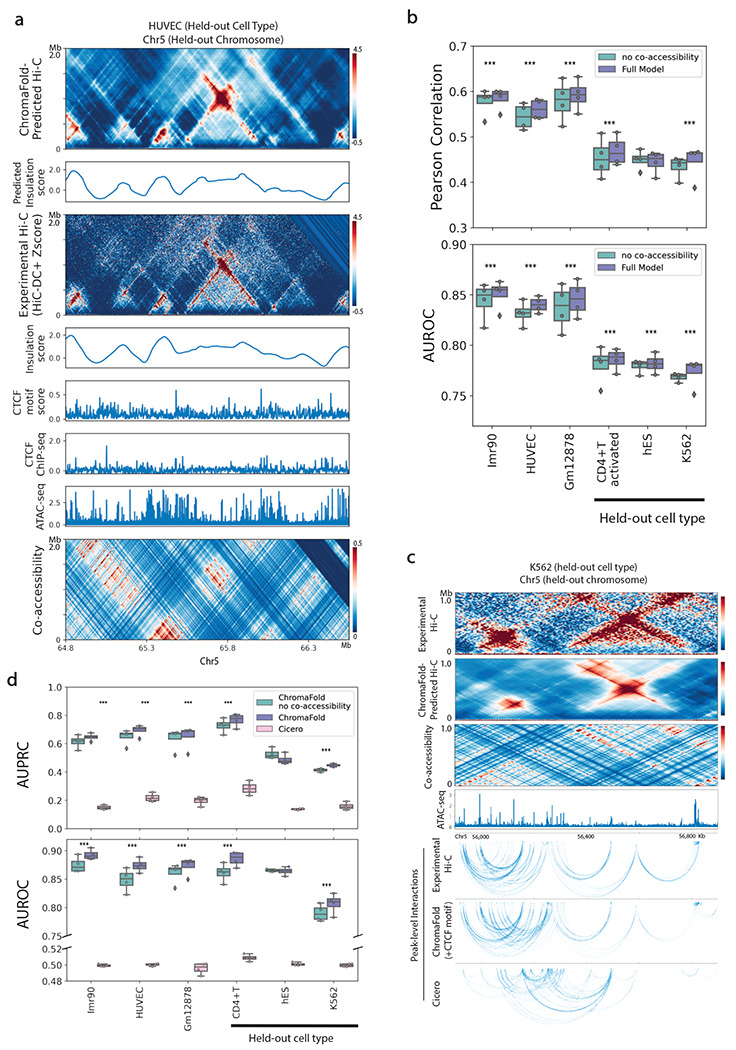
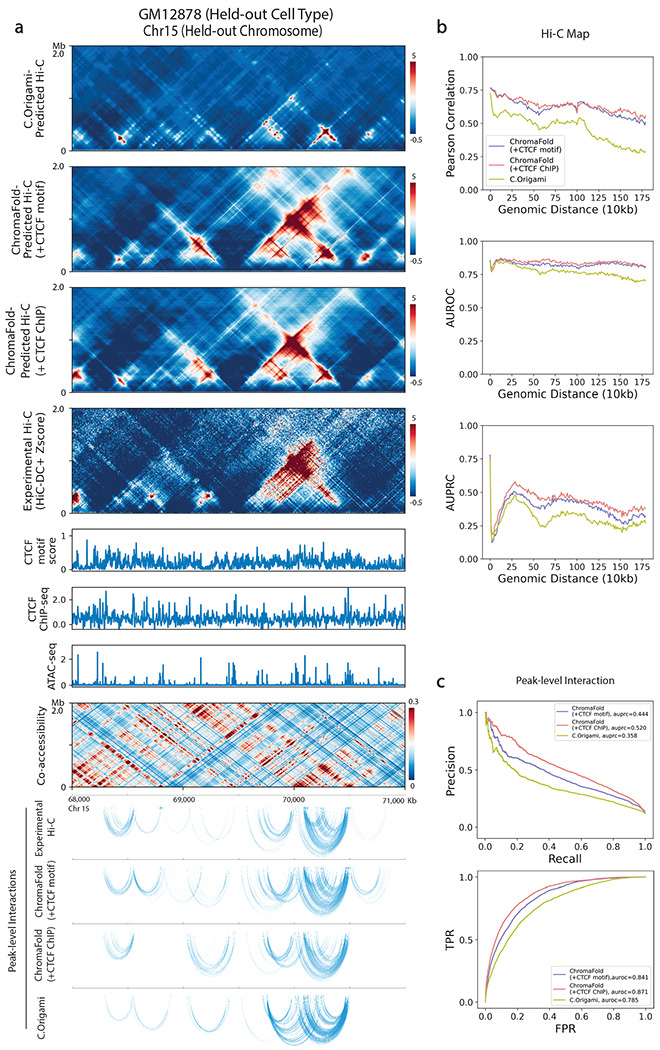
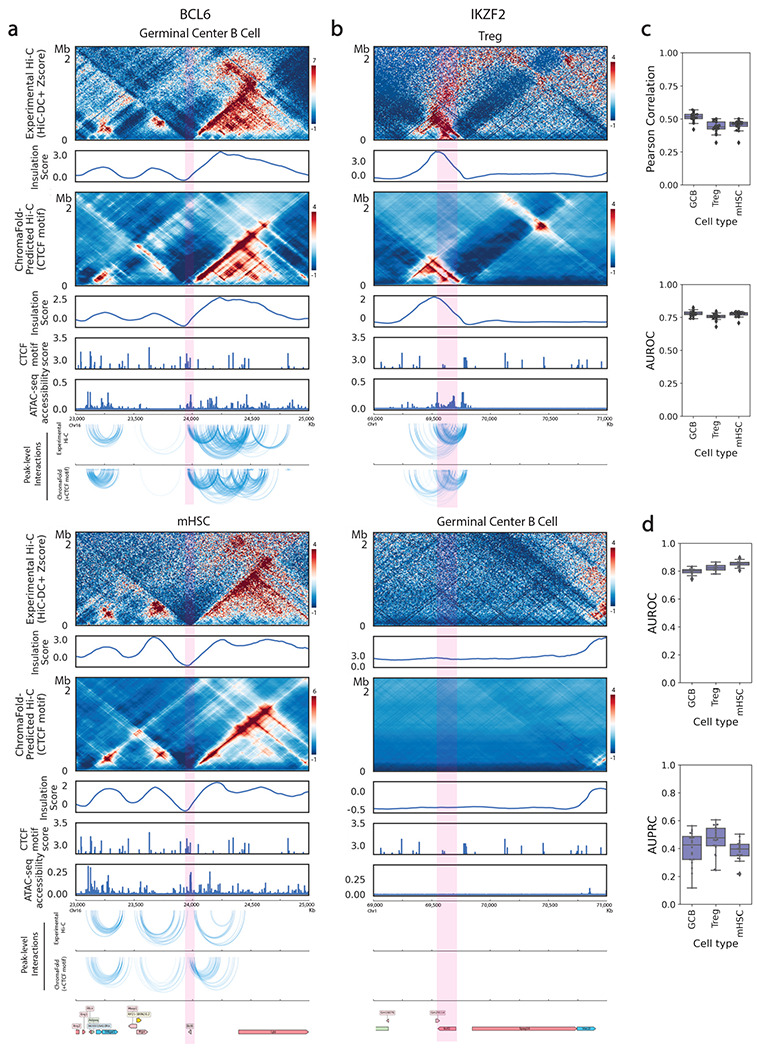
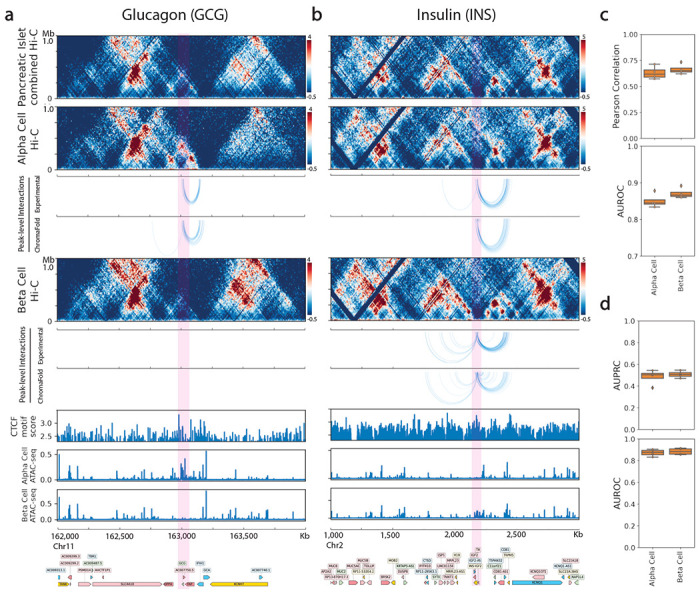
The identification of cell-type-specific 3D chromatin interactions between regulatory elements can help to decipher gene regulation and to interpret the function of disease-associated non-coding variants. However, current chromosome conformation capture (3C) technologies are unable to resolve interactions at this resolution when only small numbers of cells are available as input. We therefore present ChromaFold, a deep learning model that predicts 3D contact maps and regulatory interactions from single-cell ATAC sequencing (scATAC-seq) data alone. ChromaFold uses pseudobulk chromatin accessibility, co-accessibility profiles across metacells, and predicted CTCF motif tracks as input features and employs a lightweight architecture to enable training on standard GPUs. Once trained on paired scATAC-seq and Hi-C data in human cell lines and tissues, ChromaFold can accurately predict both the 3D contact map and peak-level interactions across diverse human and mouse test cell types. In benchmarking against a recent deep learning method that uses bulk ATAC-seq, DNA sequence, and CTCF ChIP-seq to make cell-type-specific predictions, ChromaFold yields superior prediction performance when including CTCF ChIP-seq data as an input and comparable performance without. Finally, fine-tuning ChromaFold on paired scATAC-seq and Hi-C in a complex tissue enables deconvolution of chromatin interactions across cell subpopulations. ChromaFold thus achieves state-of-the-art prediction of 3D contact maps and regulatory interactions using scATAC-seq alone as input data, enabling accurate inference of cell-type-specific interactions in settings where 3C-based assays are infeasible.
Conflict of interest statement
C.S.L. is an SAB member and co-inventor of IP with Episteme Prognostics, unrelated to the current study. M.G.K. is a SAB member of 858 Therapeutics and received honorarium from Kumquat, AstraZeneca and Consultancy with Transition Bio. A.D.V is an SAB member of Arima Genomics. A.Y.R. is an SAB member and has equity in Sonoma Biotherapeutics, Santa Ana Bio, RAPT Therapeutics and Vedanta Biosciences. He is an SEB member of Amgen and BioInvent and is a co-inventor or has IP licensed to Takeda that is unrelated to the content of the present study. A.M. has research funding from Janssen, Epizyme and Daiichi Sankyo. A.M. has consulted for Exo Therapeutics, Treeline Biosciences, Astra Zeneca. The remaining authors declare no competing interests.
Figures
References
-
- Krijger P. H. L. & De Laat W. Regulation of disease-associated gene expression in the 3D genome. Nature reviews Molecular cell biology 17, 771–782 (2016). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases