

This is a preprint.
Spatial and single-nucleus transcriptomic analysis of genetic and sporadic forms of Alzheimer's Disease
- PMID: 37546983
- PMCID: PMC10402031
- DOI: 10.1101/2023.07.24.550282
Spatial and single-nucleus transcriptomic analysis of genetic and sporadic forms of Alzheimer's Disease
Update in
-
Spatial and single-nucleus transcriptomic analysis of genetic and sporadic forms of Alzheimer's disease.Nat Genet. 2024 Dec;56(12):2704-2717. doi: 10.1038/s41588-024-01961-x. Epub 2024 Nov 22. Nat Genet. 2024. PMID: 39578645 Free PMC article.
Abstract
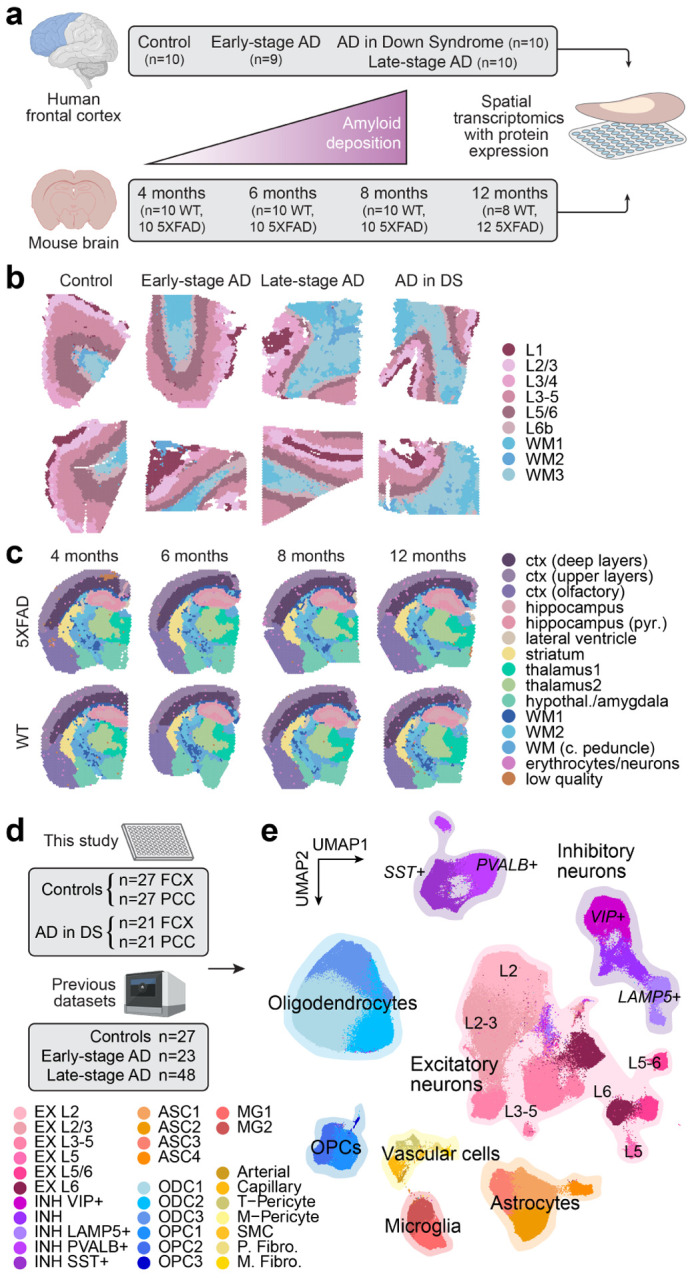
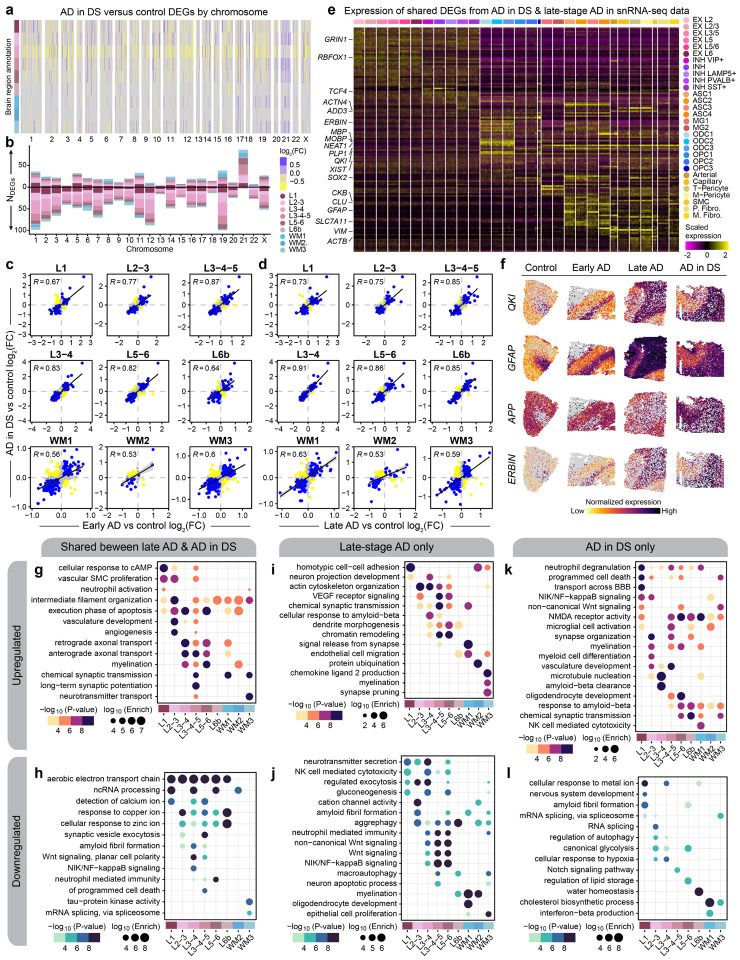
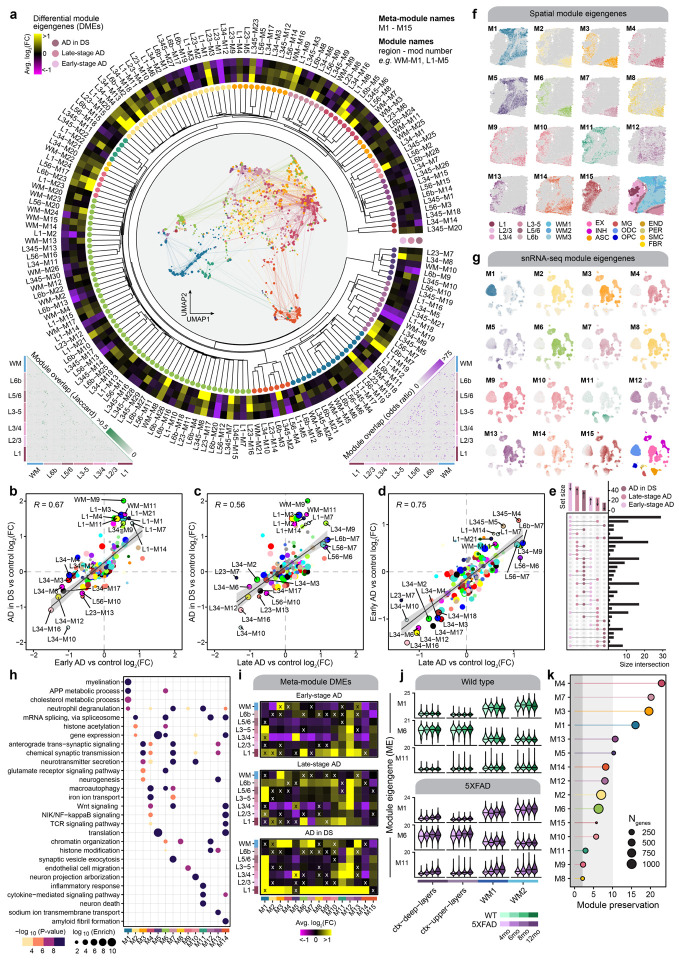
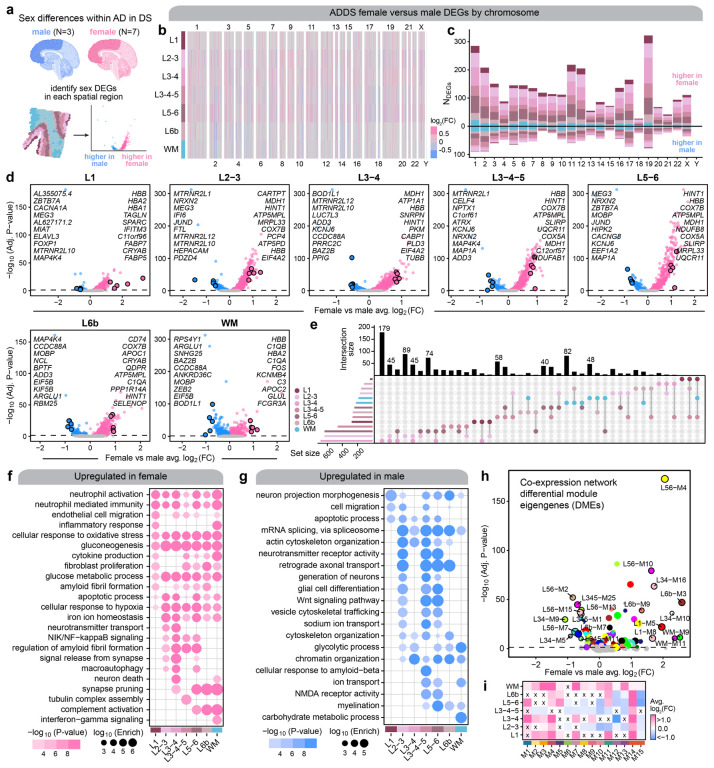
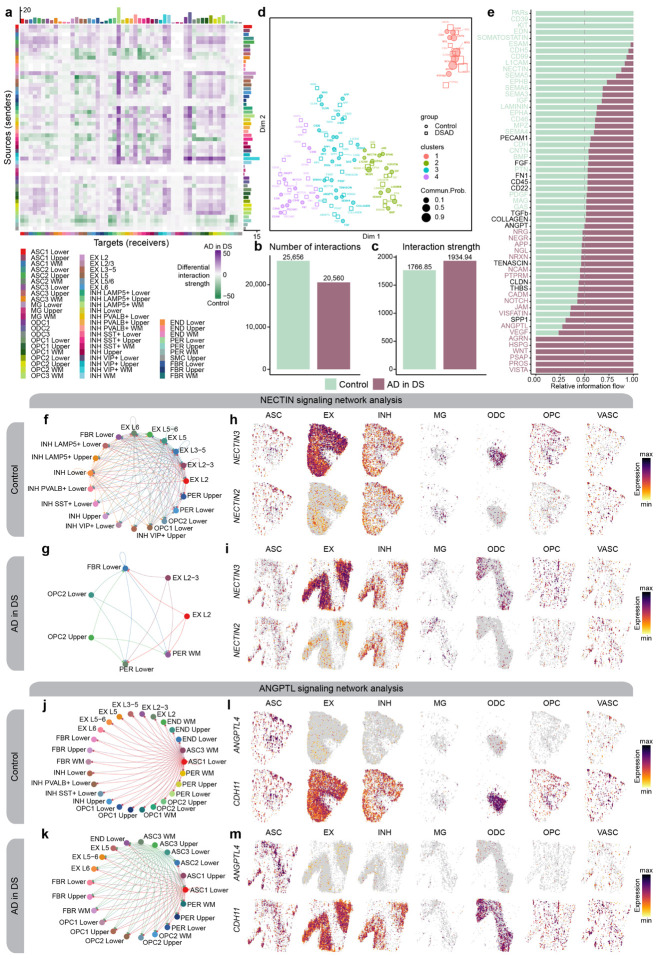
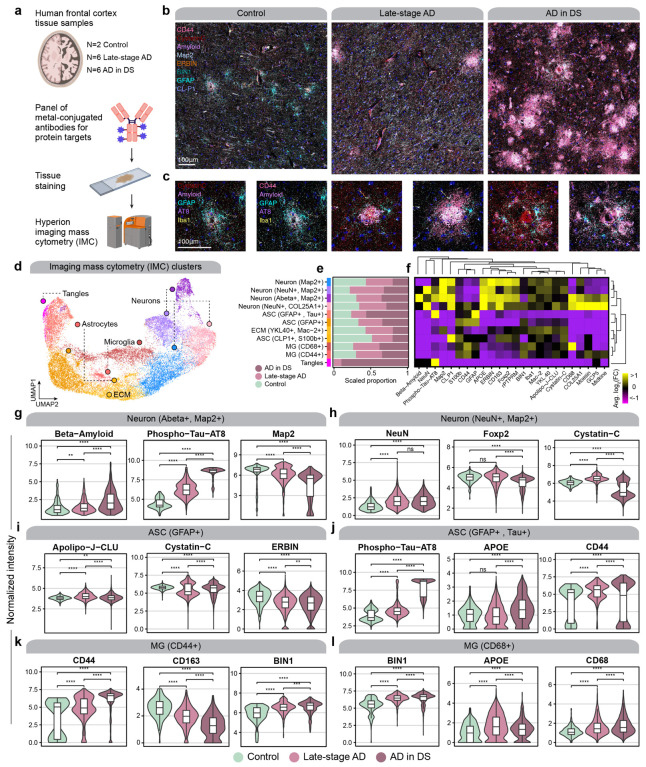
The pathogenesis of Alzheimer's disease (AD) depends on environmental and heritable factors, with remarkable differences evident between individuals at the molecular level. Here we present a transcriptomic survey of AD using spatial transcriptomics (ST) and single-nucleus RNA-seq in cortical samples from early-stage AD, late-stage AD, and AD in Down Syndrome (AD in DS) donors. Studying AD in DS provides an opportunity to enhance our understanding of the AD transcriptome, potentially bridging the gap between genetic mouse models and sporadic AD. Our analysis revealed spatial and cell-type specific changes in disease, with broad similarities in these changes between sAD and AD in DS. We performed additional ST experiments in a disease timecourse of 5xFAD and wildtype mice to facilitate cross-species comparisons. Finally, amyloid plaque and fibril imaging in the same tissue samples used for ST enabled us to directly link changes in gene expression with accumulation and spread of pathology.
Figures


References
-
- BRAIN Initiative Cell Census Network (BICCN), BRAIN Initiative Cell Census Network (BICCN) Corresponding authors, Callaway Edward M, Dong Hong-Wei, Ecker Joseph R, Hawrylycz Michael J, Huang Z Josh, Ed Lein S, Ngai John, Osten Pavel, et al. A multimodal cell census and atlas of the mammalian primary motor cortex. Nature, 598(7879):86–102, 2021. ISSN 0028–0836. doi: 10.1038/s41586-021-03950-0. - DOI - PMC - PubMed
-
- Lake Blue B., Ai Rizi, Kaeser Gwendolyn E., Salathia Neeraj S., Yung Yun C., Liu Rui, Wildberg Andre, Gao Derek, Fung Ho-Lim, Chen Song, et al. Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science, 352(6293):1586–1590, 2016. ISSN 0036–8075. doi: 10.1126/science.aaf1204. - DOI - PMC - PubMed
-
- Darmanis Spyros, Sloan Steven A., Zhang Ye, Enge Martin, Caneda Christine, Shuer Lawrence M., Hayden Gephart Melanie G., Barres Ben A., and Quake Stephen R.. A survey of human brain transcriptome diversity at the single cell level. Proceedings of the National Academy of Sciences, 112(23):7285–7290, 2015. ISSN 0027–8424. doi: 10.1073/pnas.1507125112. - DOI - PMC - PubMed
-
- Hodge Rebecca D., Bakken Trygve E., Miller Jeremy A., Smith Kimberly A., Barkan Eliza R., Graybuck Lucas T., Close Jennie L., Long Brian, Johansen Nelson, Penn Osnat, et al. Conserved cell types with divergent features in human versus mouse cortex. Nature, 573 (7772):61–68, 2019. ISSN 0028–0836. doi: 10.1038/s41586-019-1506-7. - DOI - PMC - PubMed
-
- Tasic Bosiljka, Menon Vilas, Thuc Nghi Nguyen Tae Kyung Kim, Jarsky Tim, Yao Zizhen, Levi Boaz, Gray Lucas T, Sorensen Staci A, Dolbeare Tim, et al. Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nature Neuroscience, 19(2):335–346, 2016. ISSN 1097–6256. doi: 10.1038/nn.4216. - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous