

This is a preprint.
PMS1 as a target for splice modulation to prevent somatic CAG repeat expansion in Huntington's disease
- PMID: 37547003
- PMCID: PMC10402039
- DOI: 10.1101/2023.07.25.550489
PMS1 as a target for splice modulation to prevent somatic CAG repeat expansion in Huntington's disease
Update in
-
Splice modulators target PMS1 to reduce somatic expansion of the Huntington's disease-associated CAG repeat.Nat Commun. 2024 Apr 12;15(1):3182. doi: 10.1038/s41467-024-47485-0. Nat Commun. 2024. PMID: 38609352 Free PMC article.
Abstract
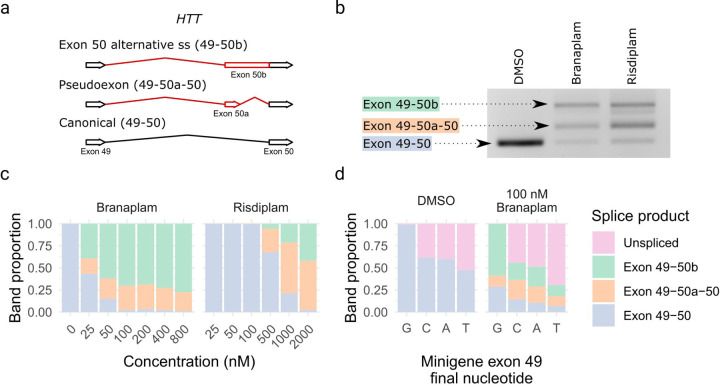
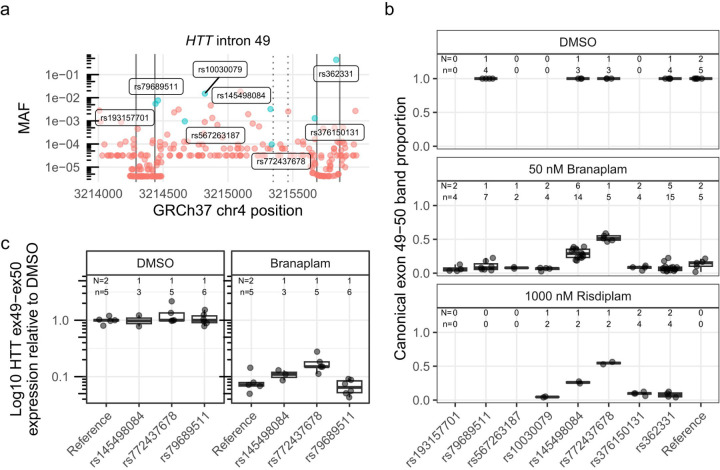
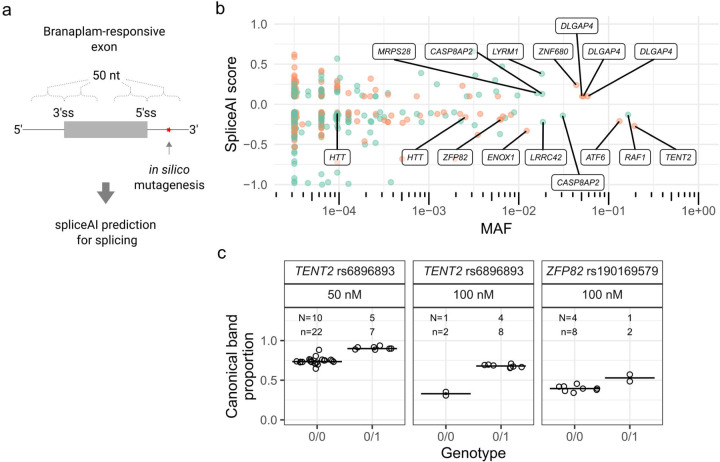
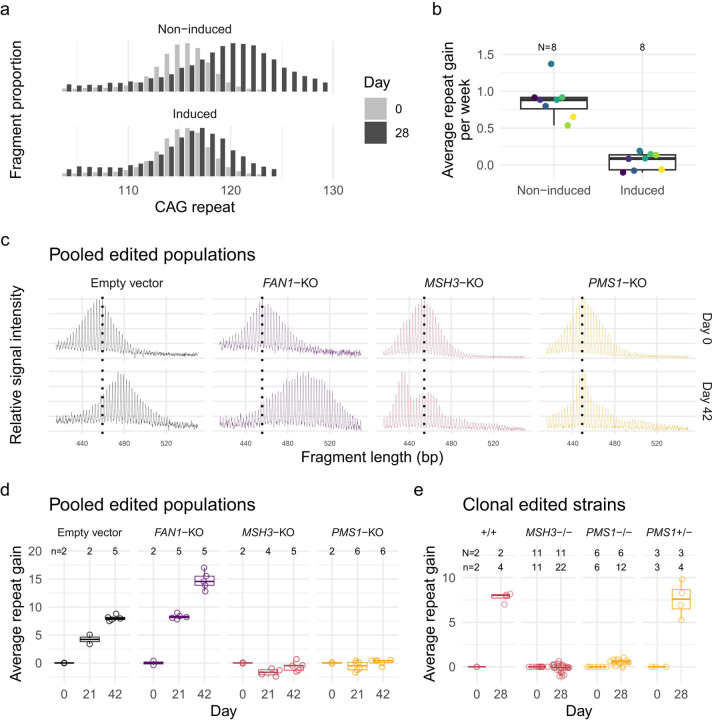
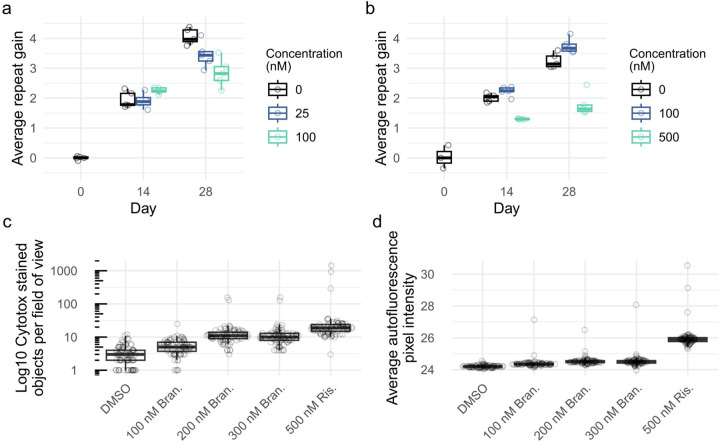
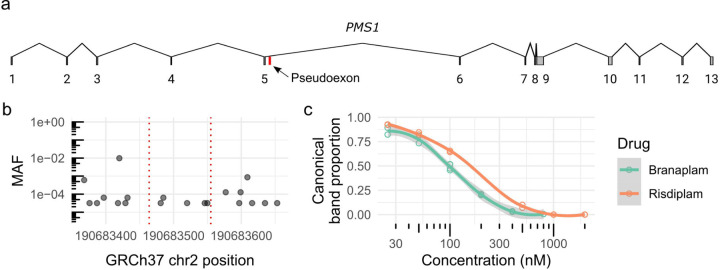
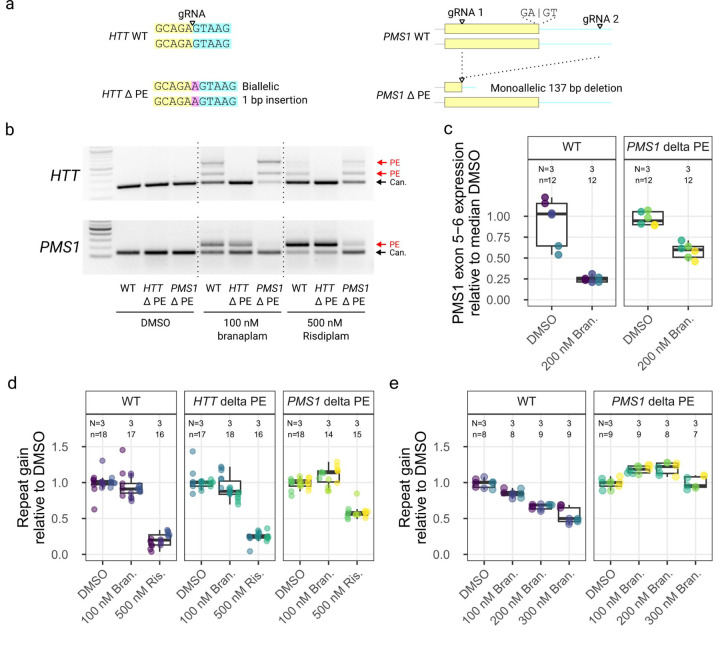
Huntington's disease (HD) is a dominantly inherited neurodegenerative disorder whose motor, cognitive, and behavioral manifestations are caused by an expanded, somatically unstable CAG repeat in the first exon of HTT that lengthens a polyglutamine tract in huntingtin. Genome-wide association studies (GWAS) have revealed DNA repair genes that influence the age-at-onset of HD and implicate somatic CAG repeat expansion as the primary driver of disease timing. To prevent the consequent neuronal damage, small molecule splice modulators (e.g., branaplam) that target HTT to reduce the levels of huntingtin are being investigated as potential HD therapeutics. We found that the effectiveness of the splice modulators can be influenced by genetic variants, both at HTT and other genes where they promote pseudoexon inclusion. Surprisingly, in a novel hTERT-immortalized retinal pigment epithelial cell (RPE1) model for assessing CAG repeat instability, these drugs also reduced the rate of HTT CAG expansion. We determined that the splice modulators also affect the expression of the mismatch repair gene PMS1, a known modifier of HD age-at-onset. Genome editing at specific HTT and PMS1 sequences using CRISPR-Cas9 nuclease confirmed that branaplam suppresses CAG expansion by promoting the inclusion of a pseudoexon in PMS1, making splice modulation of PMS1 a potential strategy for delaying HD onset. Comparison with another splice modulator, risdiplam, suggests that other genes affected by these splice modulators also influence CAG instability and might provide additional therapeutic targets.
Conflict of interest statement
Competing interests J.F.G. and V.C.W. were founding scientific advisory board members with a financial interest in Triplet Therapeutics Inc. Their financial interests were reviewed and are managed by Massachusetts General Hospital (MGH) and Mass General Brigham (MGB) in accordance with their conflict of interest policies. J.F.G. consults for Transine Therapeutics, Inc. and has previously provided paid consulting services to Wave Therapeutics USA Inc., Biogen Inc. and Pfizer Inc. V.C.W. is a scientific advisory board member of LoQus23 Therapeutics Ltd. and has provided paid consulting services to Acadia Pharmaceuticals Inc., Alnylam Inc., Biogen Inc. and Passage Bio. R.M.P. and V.C.W. have received research support from Pfizer Inc. B.P.K. is a consultant for EcoR1 capital and Curie.Bio, and is an advisor to Acrigen Biosciences, Life Edit Therapeutics and Prime Medicine. B.P.K. has a financial interest in Prime Medicine, Inc., a company developing therapeutic CRISPR-Cas technologies for gene editing. B.P.K.’s interests were reviewed and are managed by MGH and MGB in accordance with their conflict-of-interest policies. J-M.L. consults for Life Edit Therapeutics and serves on the scientific advisory board of GenEdit Inc. E.M. is inventor on an International Patent Application Number PCT/US2021/012103, assigned to Massachusetts General Hospital and PTC Therapeutics entitled “RNA Splicing Modulation” related to use of BPN-15477 in modulating splicing.
Figures
References
-
- MacDonald M. E. et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 72, 971–983 (1993). - PubMed
-
- Kennedy L. et al. Dramatic tissue-specific mutation length increases are an early molecular event in Huntington disease pathogenesis. Human Molecular Genetics 12, 3359–3367 (2003). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials