

Antimicrobial resistance heterogeneity among multidrug-resistant Gram-negative pathogens: Phenotypic, genotypic, and proteomic analysis
- PMID: 37549252
- PMCID: PMC10434301
- DOI: 10.1073/pnas.2305465120
Antimicrobial resistance heterogeneity among multidrug-resistant Gram-negative pathogens: Phenotypic, genotypic, and proteomic analysis
Abstract
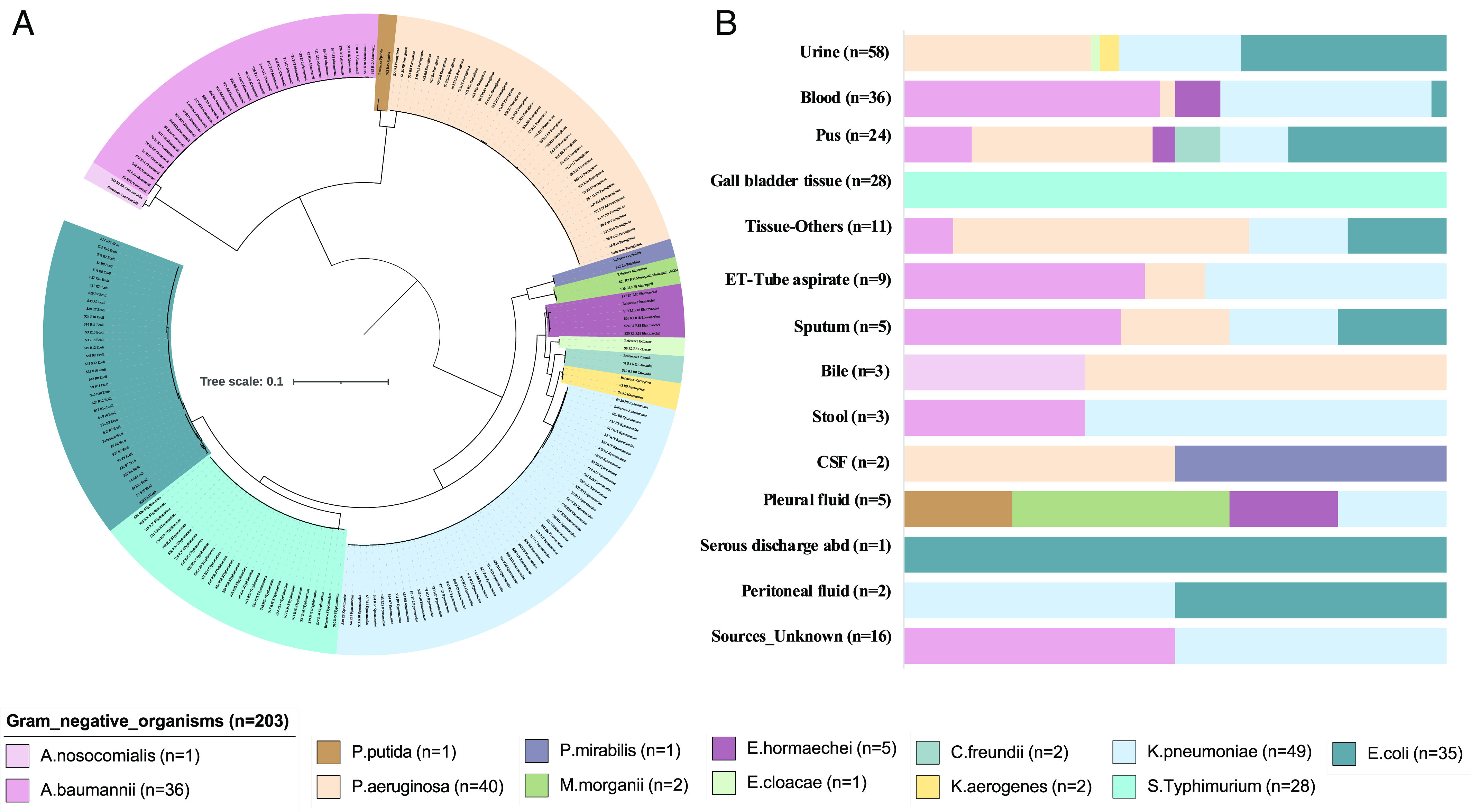
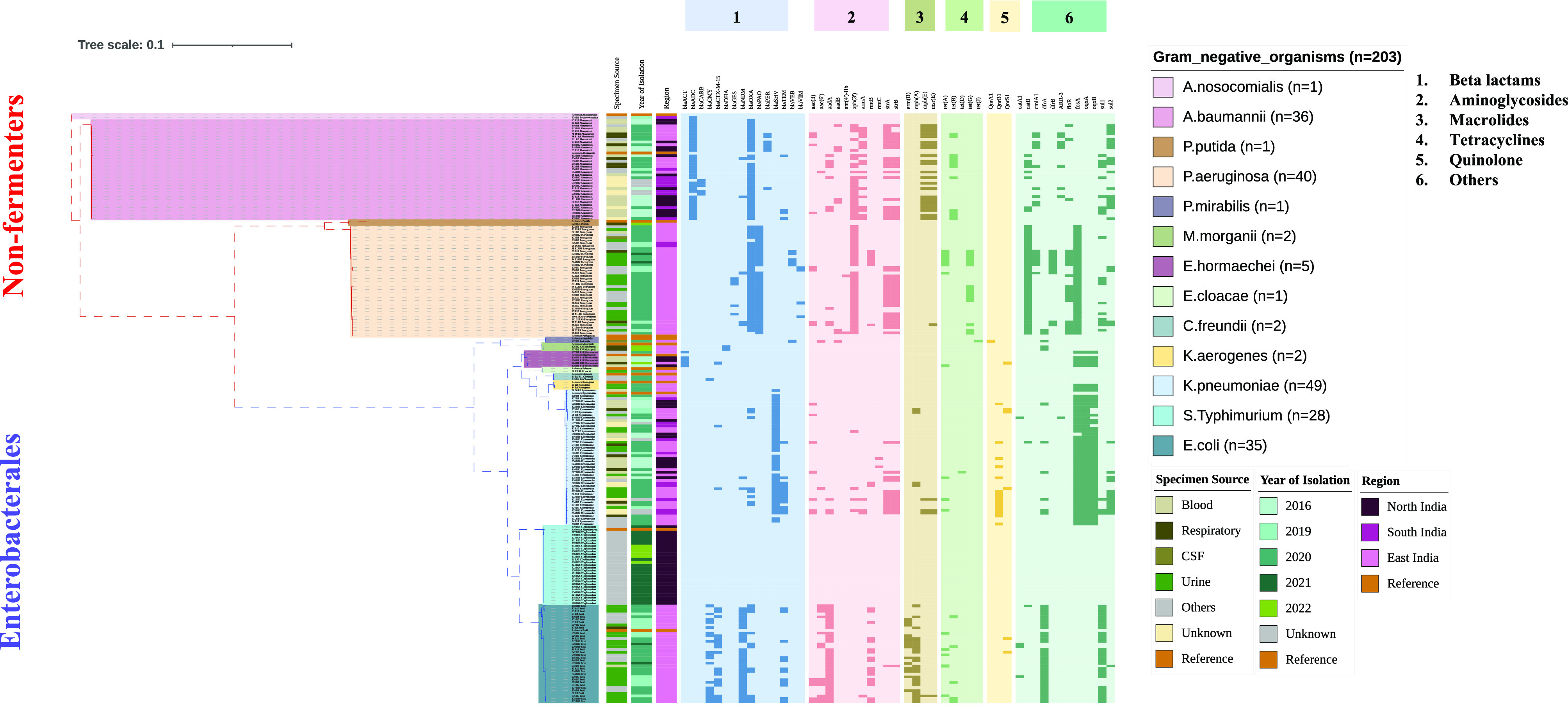
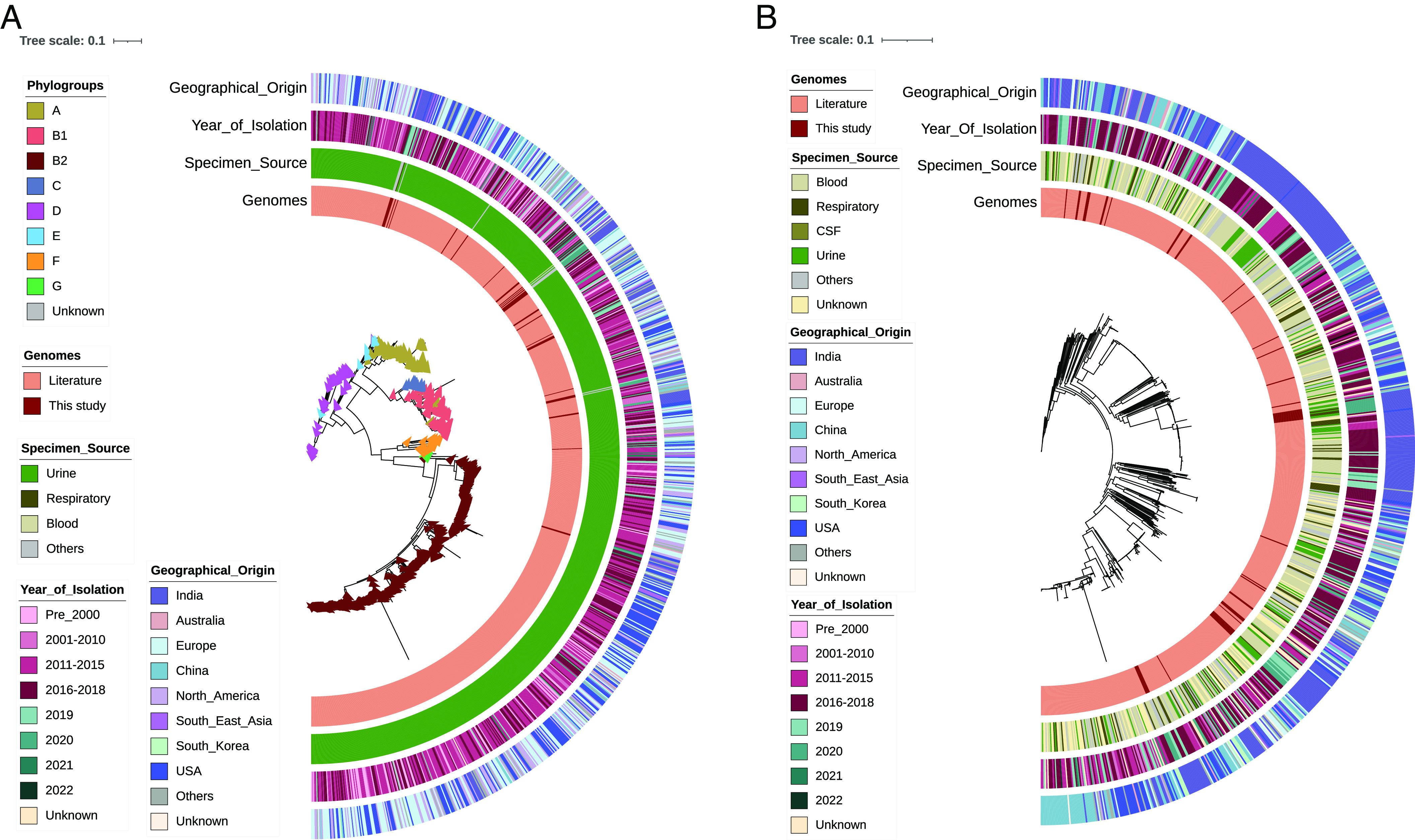
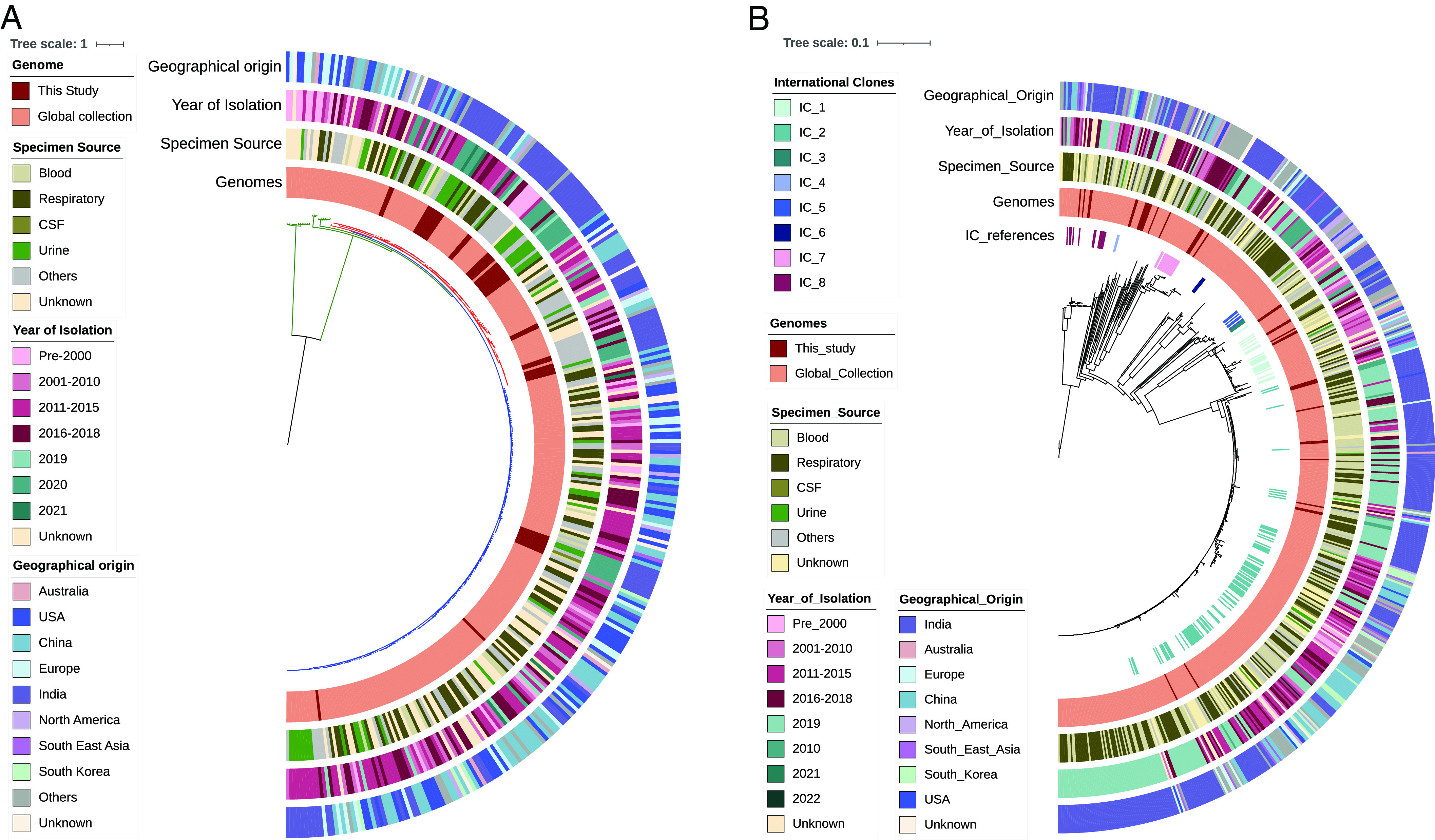
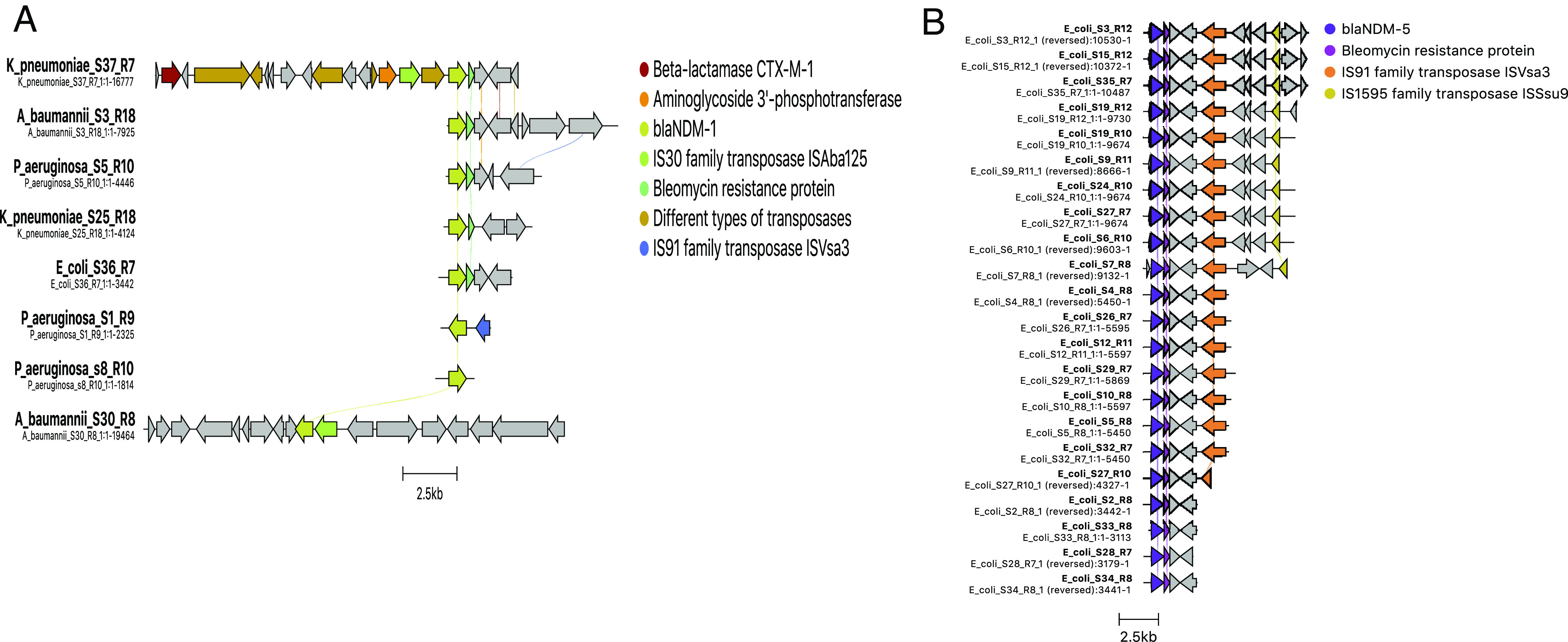
Microbes evolve rapidly by modifying their genomes through mutations or through the horizontal acquisition of mobile genetic elements (MGEs) linked with fitness traits such as antimicrobial resistance (AMR), virulence, and metabolic functions. We conducted a multicentric study in India and collected different clinical samples for decoding the genome sequences of bacterial pathogens associated with sepsis, urinary tract infections, and respiratory infections to understand the functional potency associated with AMR and its dynamics. Genomic analysis identified several acquired AMR genes (ARGs) that have a pathogen-specific signature. We observed that blaCTX-M-15, blaCMY-42, blaNDM-5, and aadA(2) were prevalent in Escherichia coli, and blaTEM-1B, blaOXA-232, blaNDM-1, rmtB, and rmtC were dominant in Klebsiella pneumoniae. In contrast, Pseudomonas aeruginosa and Acinetobacter baumannii harbored blaVEB, blaVIM-2, aph(3'), strA/B, blaOXA-23, aph(3') variants, and amrA, respectively. Regardless of the type of ARG, the MGEs linked with ARGs were also pathogen-specific. The sequence type of these pathogens was identified as high-risk international clones, with only a few lineages being predominant and region-specific. Whole-cell proteome analysis of extensively drug-resistant K. pneumoniae, A. baumannii, E. coli, and P. aeruginosa strains revealed differential abundances of resistance-associated proteins in the presence and absence of different classes of antibiotics. The pathogen-specific resistance signatures and differential abundance of AMR-associated proteins identified in this study should add value to AMR diagnostics and the choice of appropriate drug combinations for successful antimicrobial therapy.
Keywords: Gram-negative pathogens; antimicrobial resistance; antimicrobials; genomics; proteomics.
Conflict of interest statement
B.D. and D.K.C. have published a commentary in 2019.
Figures
References
-
- Liu G., Thomsen L. E., Olsen J. E., Antimicrobial-induced horizontal transfer of antimicrobial resistance genes in bacteria: A mini-review. J. Antimicrob. Chemother. 77, 556–567 (2022). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
