

Spatially resolved protein map of intact human cytomegalovirus virions
- PMID: 37550507
- PMCID: PMC10465357
- DOI: 10.1038/s41564-023-01433-8
Spatially resolved protein map of intact human cytomegalovirus virions
Abstract
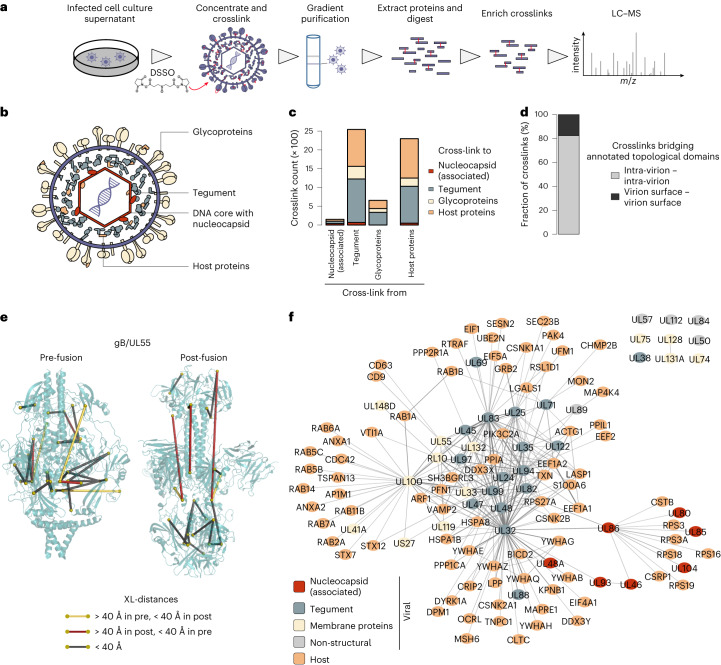
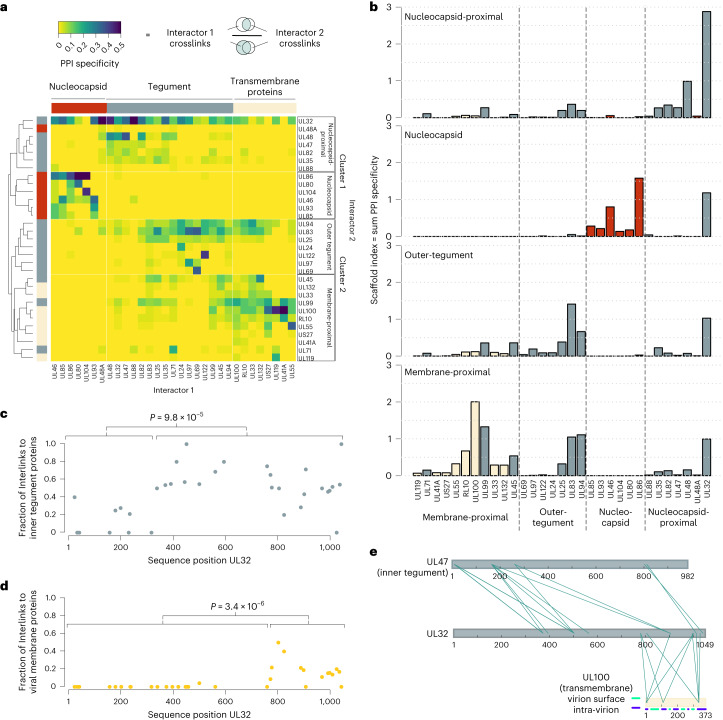
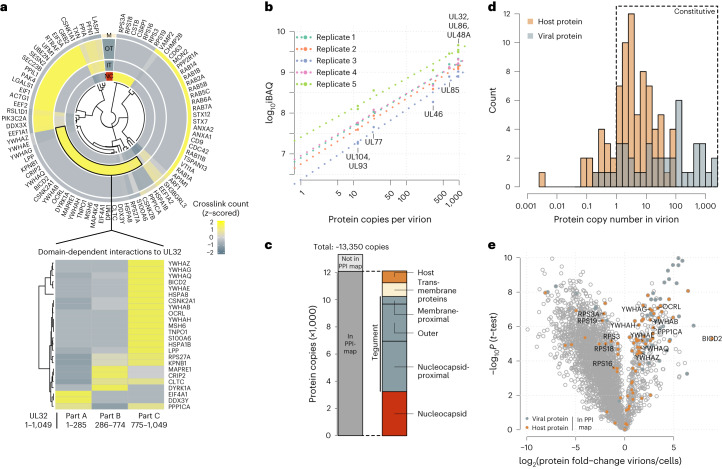
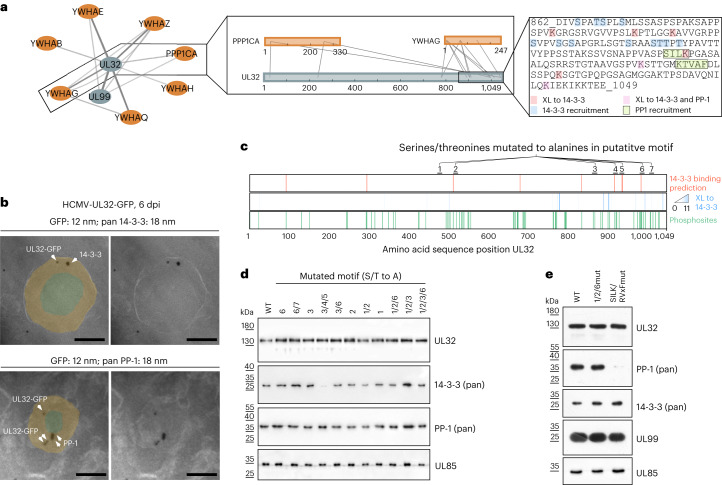
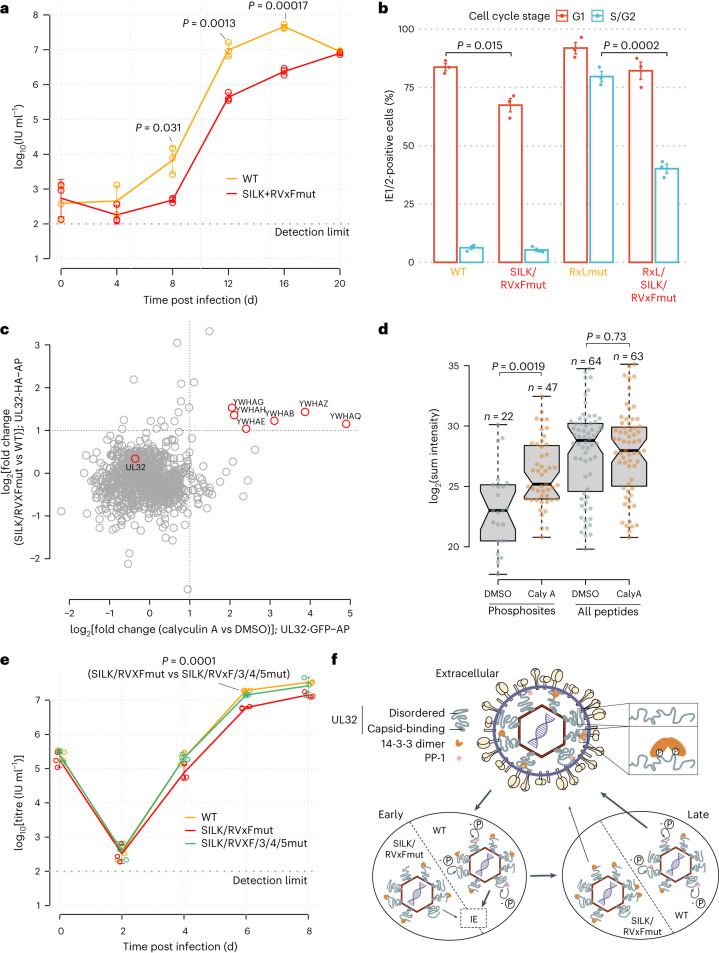
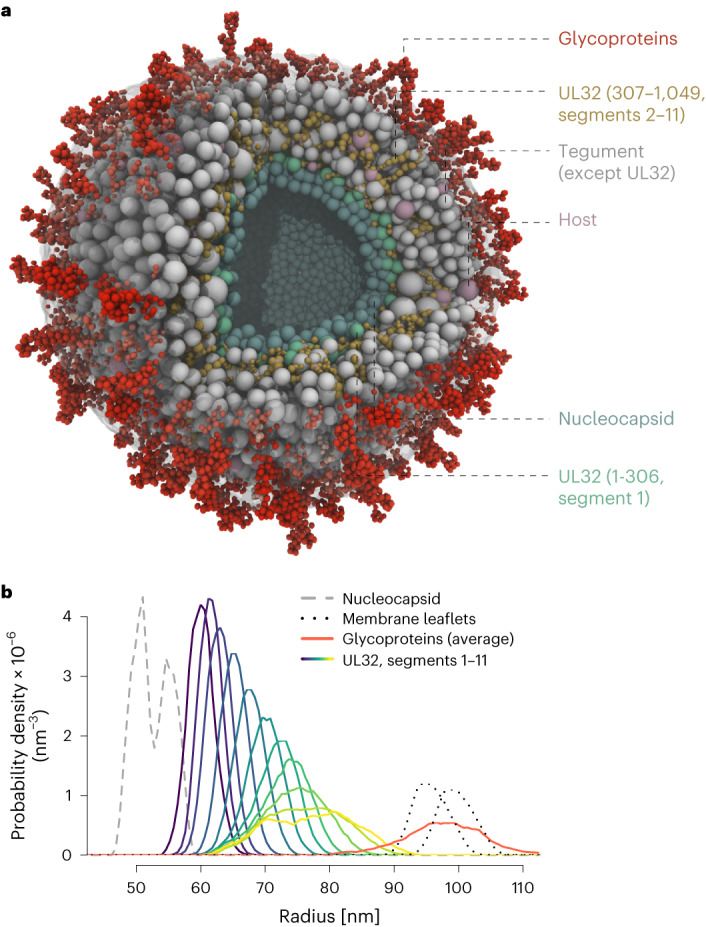
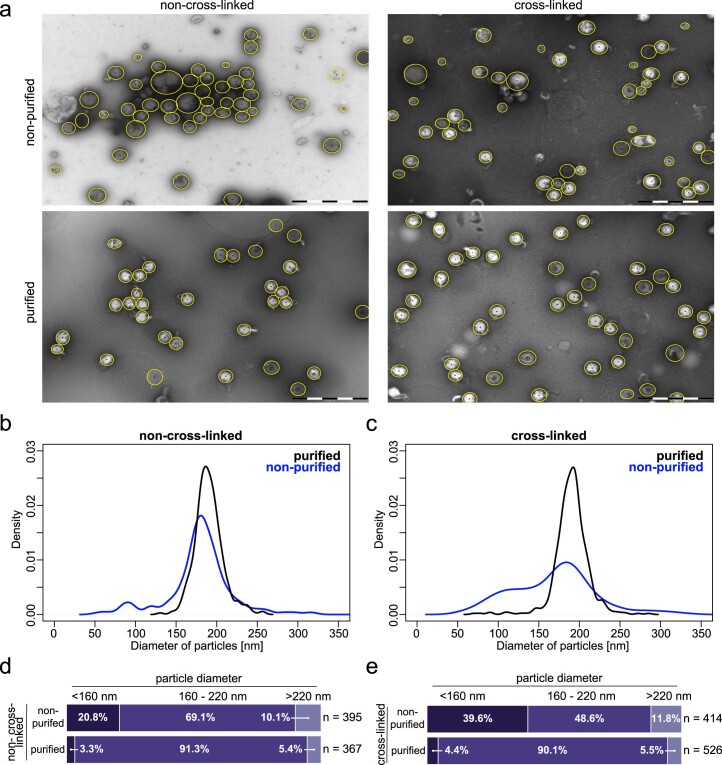
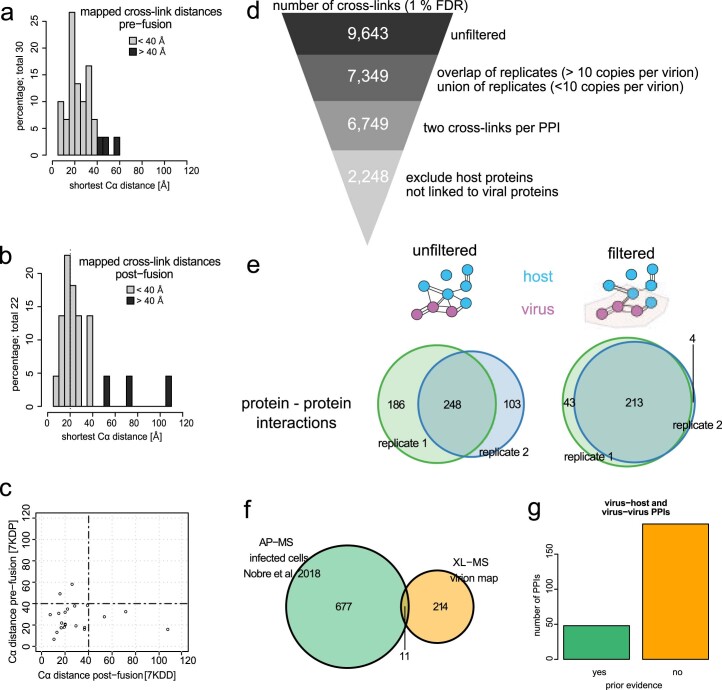
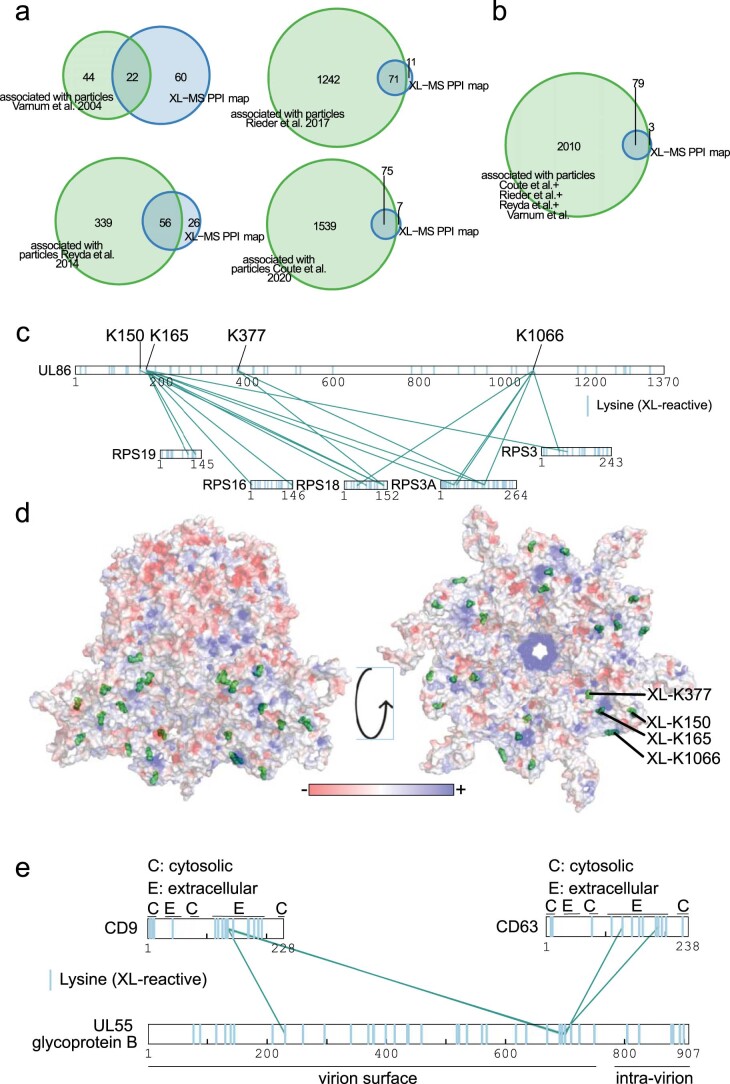
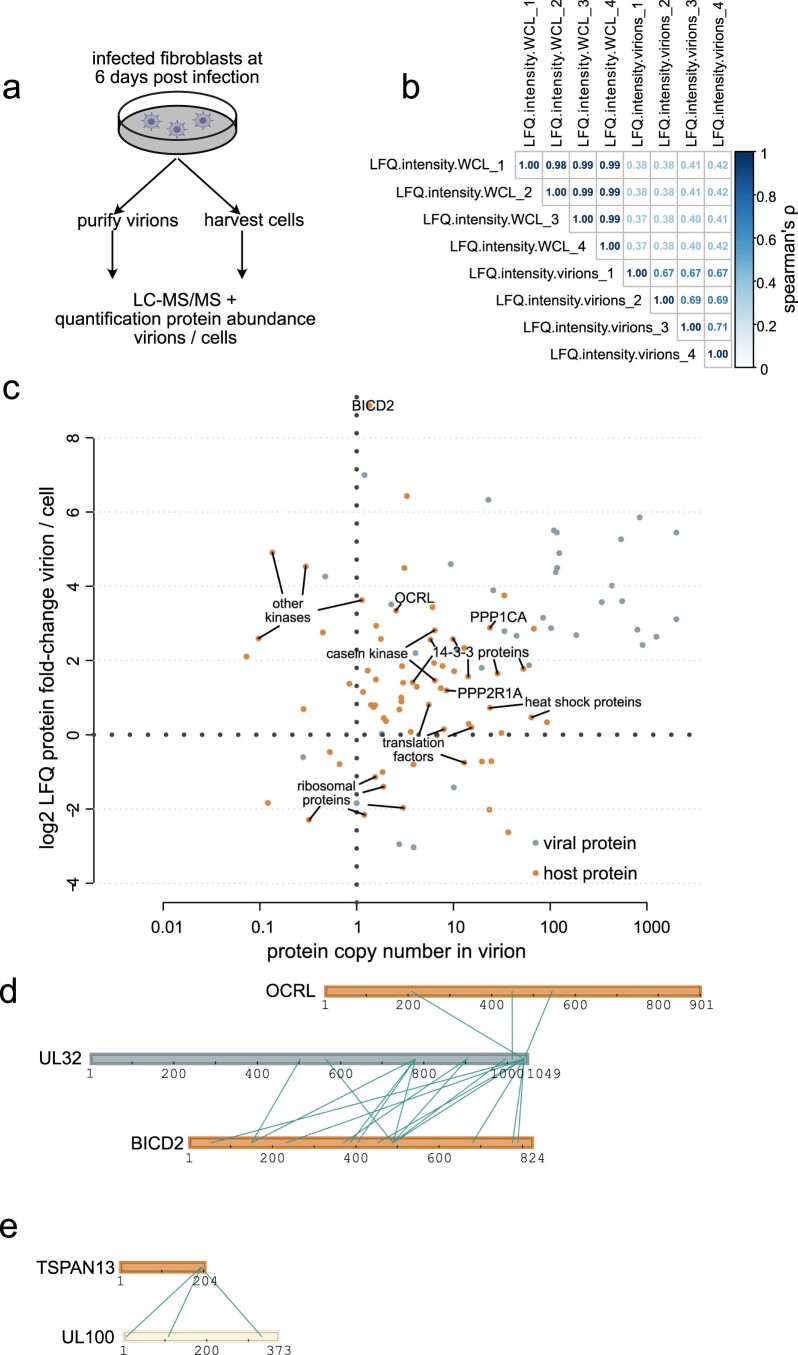
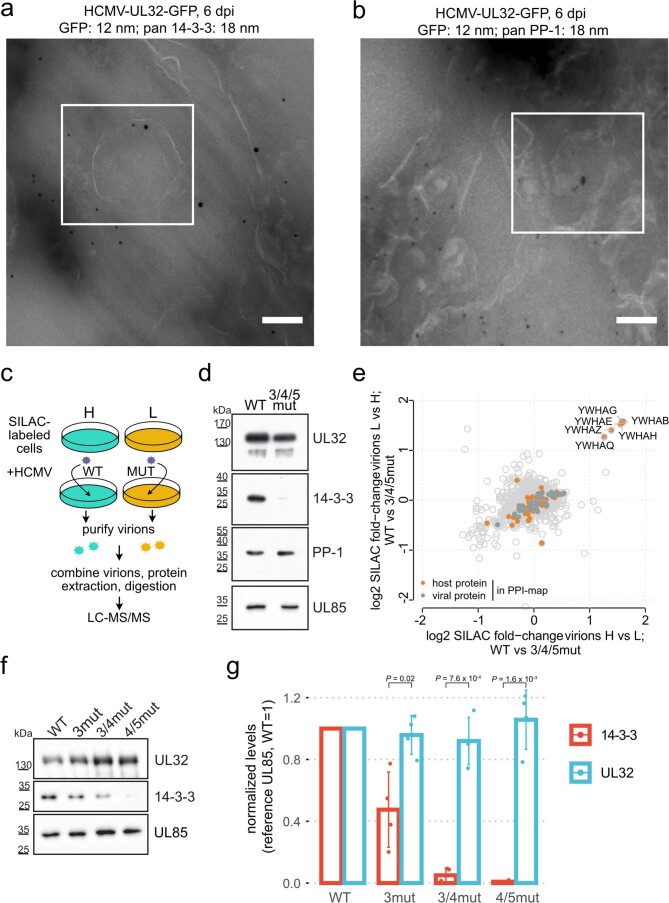
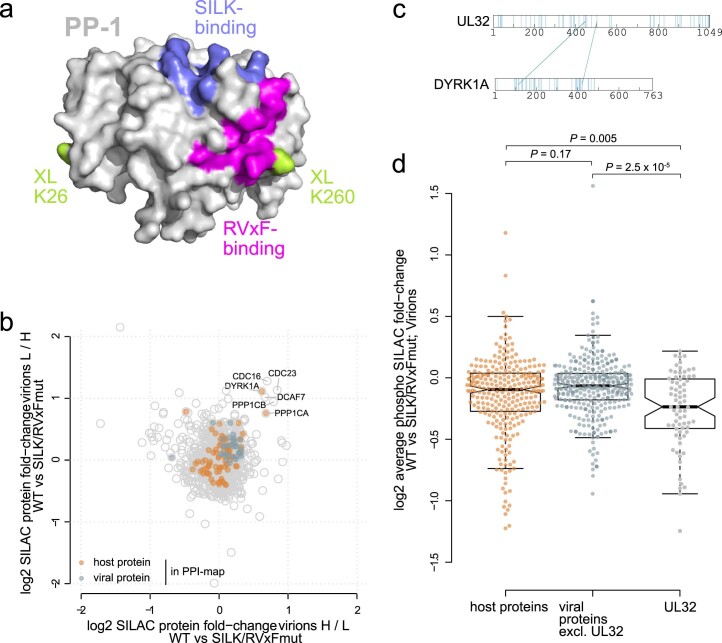
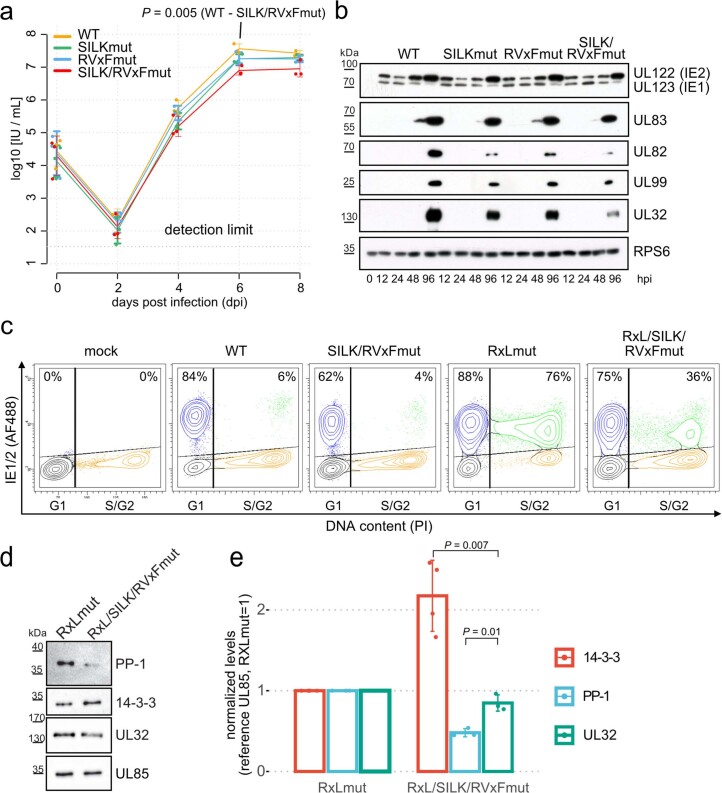
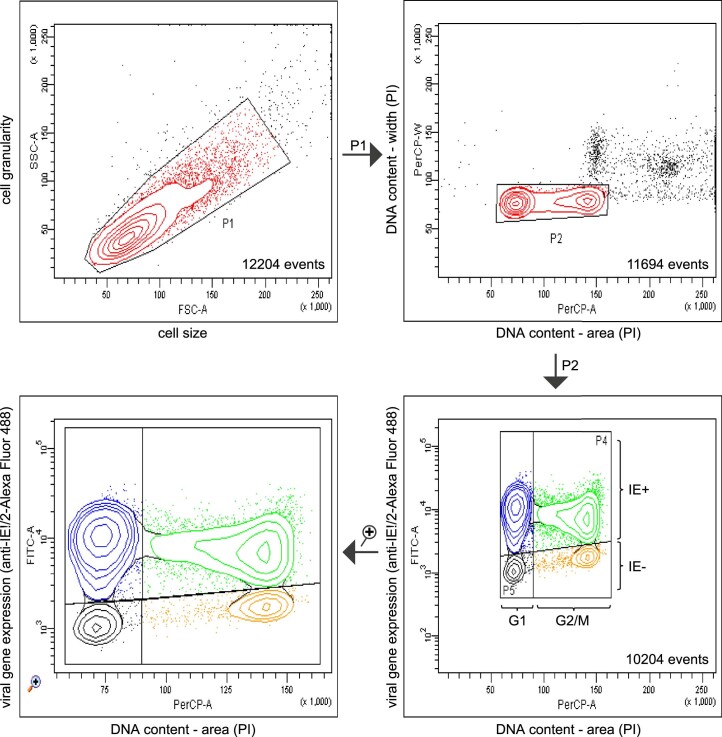
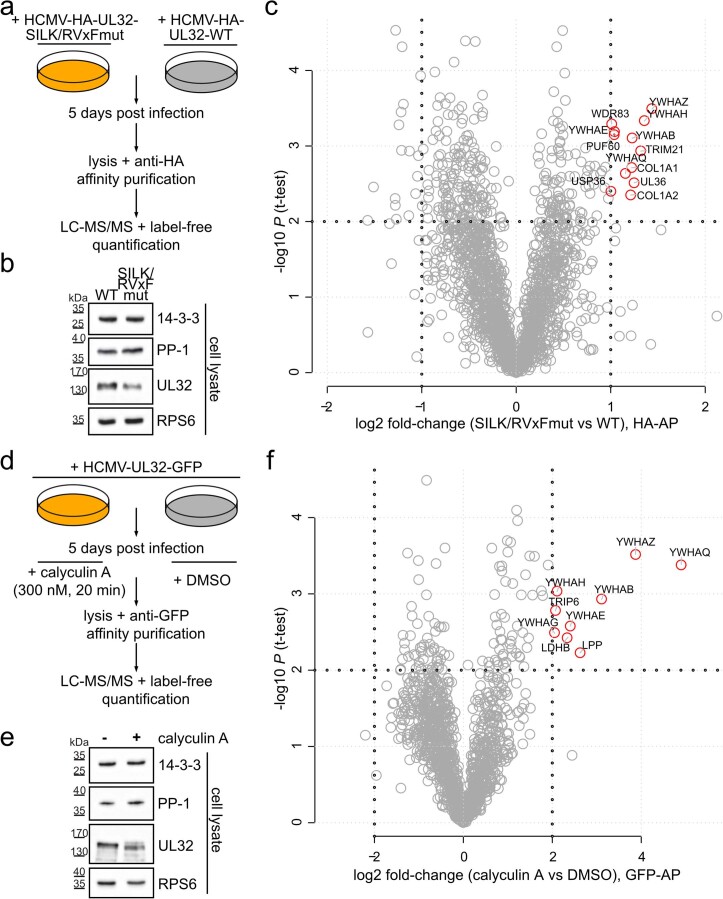
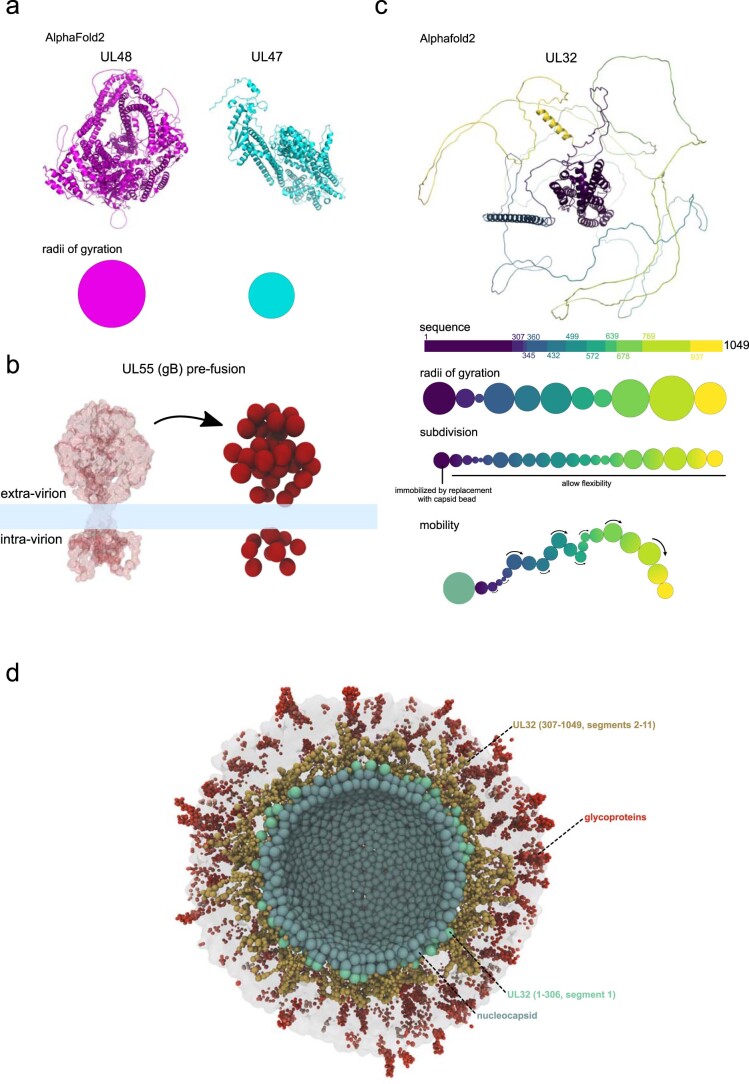
Herpesviruses assemble large enveloped particles that are difficult to characterize structurally due to their size, fragility and complex multilayered proteome with partially amorphous nature. Here we used crosslinking mass spectrometry and quantitative proteomics to derive a spatially resolved interactome map of intact human cytomegalovirus virions. This enabled the de novo allocation of 32 viral proteins into four spatially resolved virion layers, each organized by a dominant viral scaffold protein. The viral protein UL32 engages with all layers in an N-to-C-terminal radial orientation, bridging nucleocapsid to viral envelope. We observed the layer-specific incorporation of 82 host proteins, of which 39 are selectively recruited. We uncovered how UL32, by recruitment of PP-1 phosphatase, antagonizes binding to 14-3-3 proteins. This mechanism assures effective viral biogenesis, suggesting a perturbing role of UL32-14-3-3 interaction. Finally, we integrated these data into a coarse-grained model to provide global insights into the native configuration of virus and host protein interactions inside herpesvirions.
© 2023. The Author(s).
Conflict of interest statement
F.L. is a scientific advisory board member of Absea Biotechnology and VantAI. The remaining authors declare no competing interests.
Figures
References
-
- Liu, F. & Zhou, Z. H. in Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis (eds Arvin, A. et al.) Ch. 3 (Cambridge Univ. Press, 2007). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
