

Phylogenetic inference from single-cell RNA-seq data
- PMID: 37553438
- PMCID: PMC10409753
- DOI: 10.1038/s41598-023-39995-6
Phylogenetic inference from single-cell RNA-seq data
Abstract
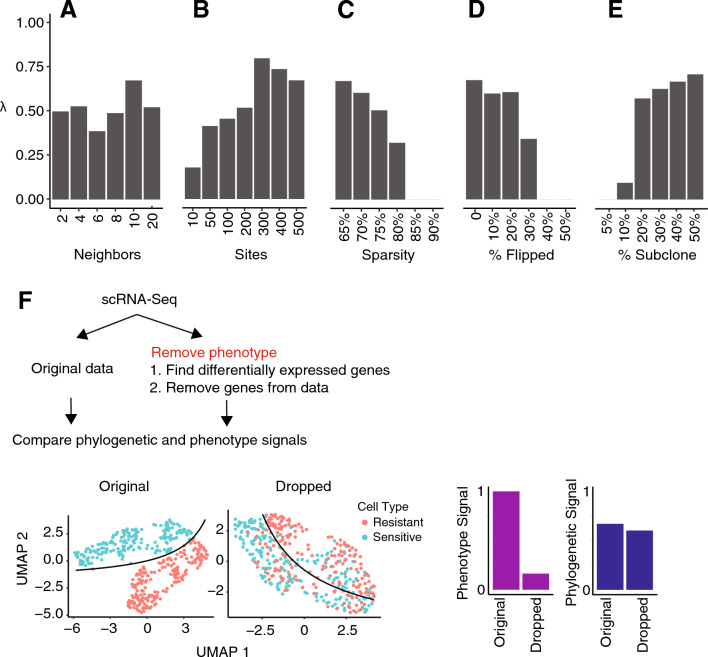
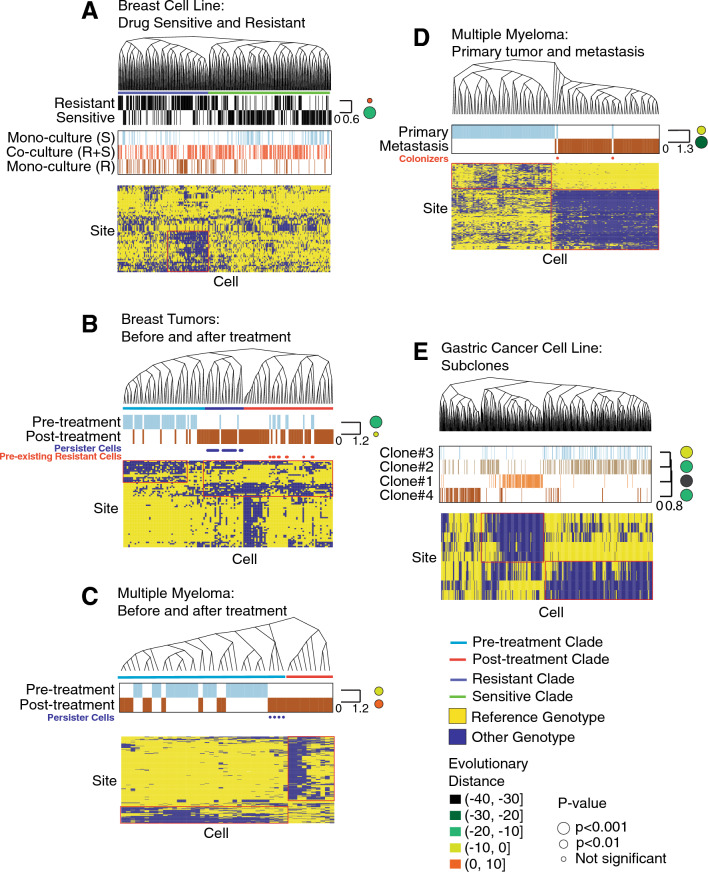
Tumors are comprised of subpopulations of cancer cells that harbor distinct genetic profiles and phenotypes that evolve over time and during treatment. By reconstructing the course of cancer evolution, we can understand the acquisition of the malignant properties that drive tumor progression. Unfortunately, recovering the evolutionary relationships of individual cancer cells linked to their phenotypes remains a difficult challenge. To address this need, we have developed PhylinSic, a method that reconstructs the phylogenetic relationships among cells linked to their gene expression profiles from single cell RNA-sequencing (scRNA-Seq) data. This method calls nucleotide bases using a probabilistic smoothing approach and then estimates a phylogenetic tree using a Bayesian modeling algorithm. We showed that PhylinSic identified evolutionary relationships underpinning drug selection and metastasis and was sensitive enough to identify subclones from genetic drift. We found that breast cancer tumors resistant to chemotherapies harbored multiple genetic lineages that independently acquired high K-Ras and β-catenin, suggesting that therapeutic strategies may need to control multiple lineages to be durable. These results demonstrated that PhylinSic can reconstruct evolution and link the genotypes and phenotypes of cells across monophyletic tumors using scRNA-Seq.
© 2023. Springer Nature Limited.
Conflict of interest statement
The authors declare no competing interests.
Figures



Similar articles
-
Can a Liquid Biopsy Detect Circulating Tumor DNA With Low-passage Whole-genome Sequencing in Patients With a Sarcoma? A Pilot Evaluation.Clin Orthop Relat Res. 2025 Jan 1;483(1):39-48. doi: 10.1097/CORR.0000000000003161. Epub 2024 Jun 21. Clin Orthop Relat Res. 2025. PMID: 38905450
-
Cost-effectiveness of using prognostic information to select women with breast cancer for adjuvant systemic therapy.Health Technol Assess. 2006 Sep;10(34):iii-iv, ix-xi, 1-204. doi: 10.3310/hta10340. Health Technol Assess. 2006. PMID: 16959170
-
A New Measure of Quantified Social Health Is Associated With Levels of Discomfort, Capability, and Mental and General Health Among Patients Seeking Musculoskeletal Specialty Care.Clin Orthop Relat Res. 2025 Apr 1;483(4):647-663. doi: 10.1097/CORR.0000000000003394. Epub 2025 Feb 5. Clin Orthop Relat Res. 2025. PMID: 39915110
-
Gonadotropin-releasing hormone (GnRH) analogues for premenstrual syndrome (PMS).Cochrane Database Syst Rev. 2025 Jun 10;6(6):CD011330. doi: 10.1002/14651858.CD011330.pub2. Cochrane Database Syst Rev. 2025. PMID: 40492482 Review.
-
Antiretrovirals for reducing the risk of mother-to-child transmission of HIV infection.Cochrane Database Syst Rev. 2011 Jul 6;(7):CD003510. doi: 10.1002/14651858.CD003510.pub3. Cochrane Database Syst Rev. 2011. PMID: 21735394
Cited by
-
Resolving tumor evolution: a phylogenetic approach.J Natl Cancer Cent. 2024 Mar 21;4(2):97-106. doi: 10.1016/j.jncc.2024.03.001. eCollection 2024 Jun. J Natl Cancer Cent. 2024. PMID: 39282584 Free PMC article. Review.
-
scGAA: a general gated axial-attention model for accurate cell-type annotation of single-cell RNA-seq data.Sci Rep. 2024 Sep 27;14(1):22308. doi: 10.1038/s41598-024-73356-1. Sci Rep. 2024. PMID: 39333739 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
