

The genome of okra (Abelmoschus esculentus) provides insights into its genome evolution and high nutrient content
- PMID: 37554345
- PMCID: PMC10405168
- DOI: 10.1093/hr/uhad120
The genome of okra (Abelmoschus esculentus) provides insights into its genome evolution and high nutrient content
Abstract
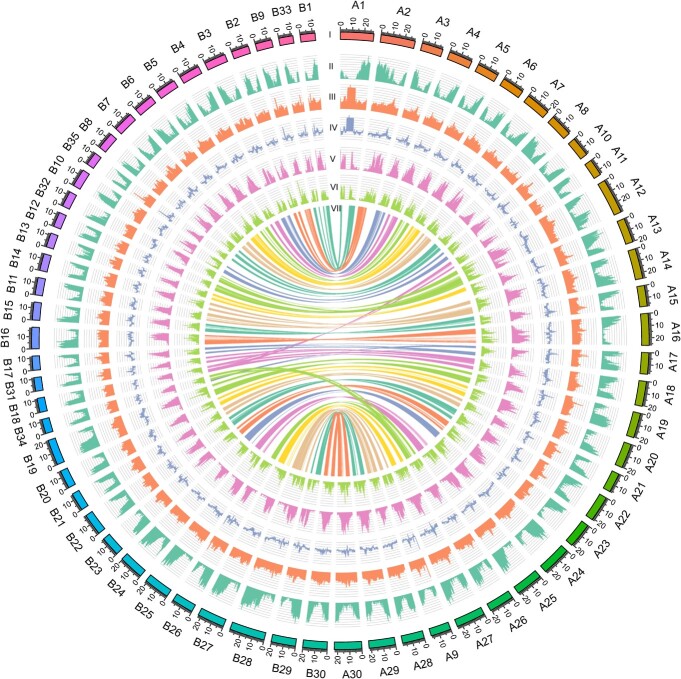
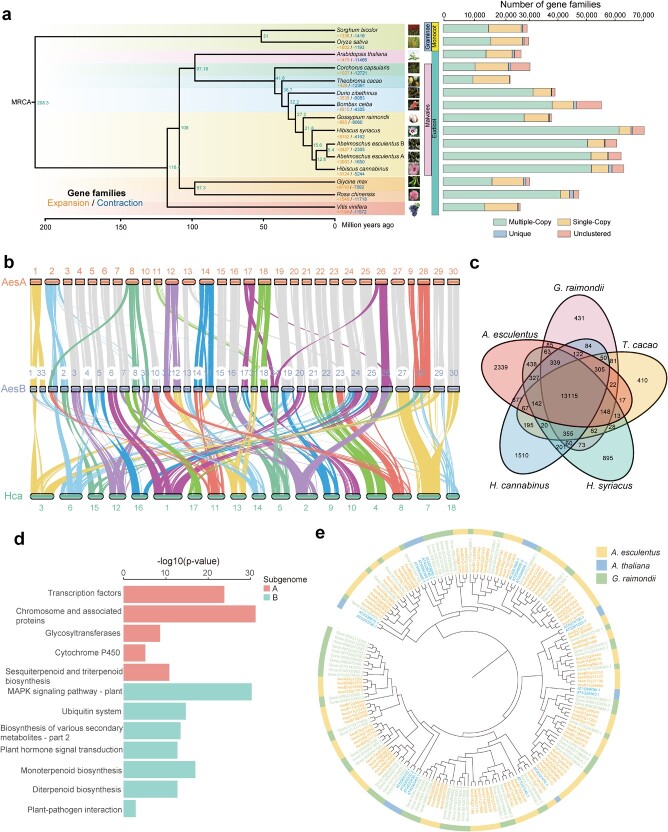
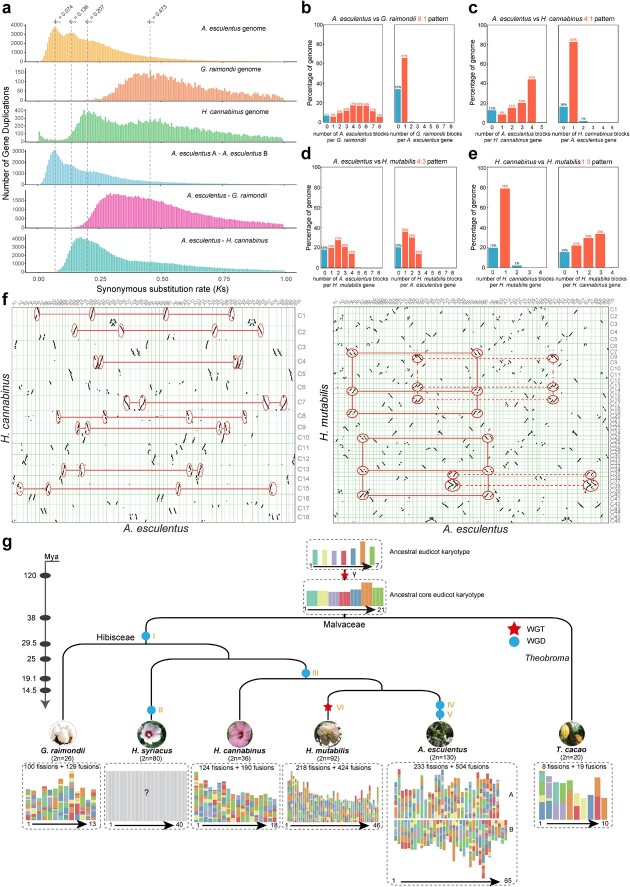
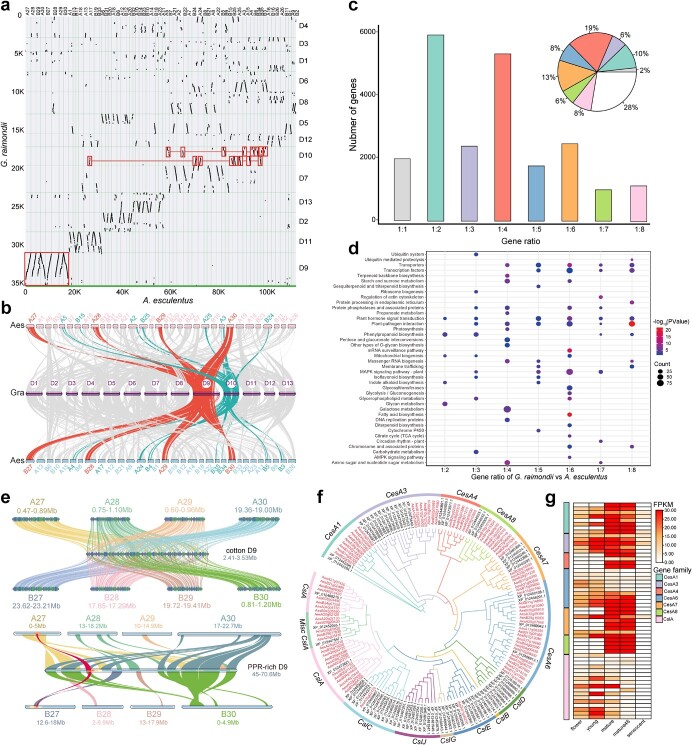
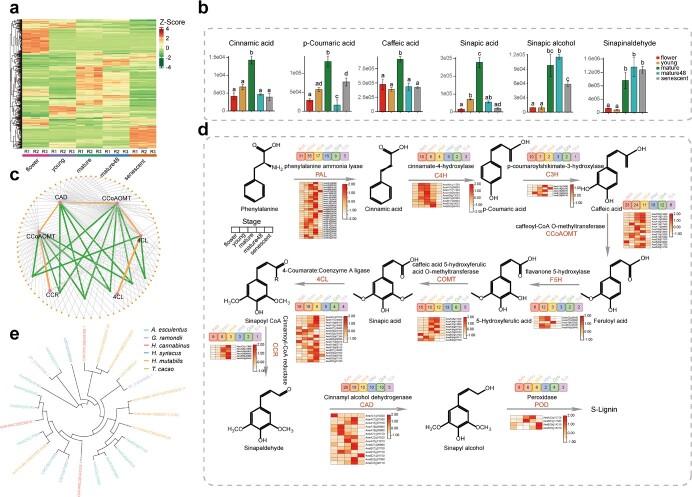
Okra (Abelmoschus esculentus) is an important vegetable crop with high nutritional value. However, the mechanism underlying its high nutrient content remains poorly understood. Here, we present a chromosome-scale genome of okra with a size of 1.19 Gb. Comparative genomics analysis revealed the phylogenetic status of A. esculentus, as well as whole-genome duplication (WGD) events that have occurred widely across the Malvaceae species. We found that okra has experienced three additional WGDs compared with the diploid cotton Gossypium raimondii, resulting in a large chromosome number (2n = 130). After three WGDs, okra has undergone extensive genomic deletions and retained substantial numbers of genes related to secondary metabolite biosynthesis and environmental adaptation, resulting in significant differences between okra and G. raimondii in the gene families related to cellulose synthesis. Combining transcriptomic and metabolomic analysis, we revealed the relationship between gene expression and metabolite content change across different okra developmental stages. Furthermore, the sinapic acid/S-lignin biosynthesis-related gene families have experienced remarkable expansion in okra, and the expression of key enzymes involved in the sinapic acid/S-lignin biosynthesis pathway vary greatly across developmental periods, which partially explains the differences in metabolite content across the different stages. Our study gains insights into the comprehensive evolutionary history of Malvaceae species and the genetic basis that underlies the nutrient content changes in okra, which will facilitate the functional study and genetic improvement of okra varieties.
© The Author(s) 2023. Published by Oxford University Press on behalf of Nanjing Agricultural University.
Conflict of interest statement
The authors have declared no competing interests.
Figures
References
-
- Islam MT. Phytochemical information and pharmacological activities of okra (Abelmoschus esculentus): a literature-based review. Phytother Res. 2019;33:72–80. - PubMed
-
- Yan T, Liu B, Wang Net al. . The flavonoids of okra insulates against oxidative stress, neuroinflammation and restores BDNF levels in Abeta(1)(−)(42) induced mouse model of Alzheimer's disease. Exp Gerontol. 2021;147:111263. - PubMed
-
- Fan S, Zhang Y, Sun Qet al. . Extract of okra lowers blood glucose and serum lipids in high-fat diet-induced obese C57BL/6 mice. J Nutr Biochem. 2014;25:702–9. - PubMed
LinkOut - more resources
Full Text Sources
