

Caveolin-1 in endothelial cells: A potential therapeutic target for atherosclerosis
- PMID: 37554846
- PMCID: PMC10405014
- DOI: 10.1016/j.heliyon.2023.e18653
Caveolin-1 in endothelial cells: A potential therapeutic target for atherosclerosis
Abstract
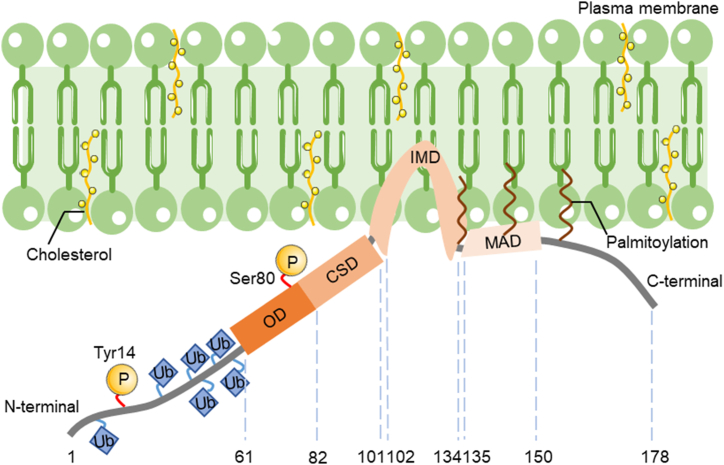
Atherosclerosis (AS) is a chronic vascular disease characterized by lipid accumulation and the activation of the inflammatory response; it remains the leading nation-wide cause of death. Early in the progression of AS, stimulation by pro-inflammatory agonists (TNF-α, LPS, and others), oxidized lipoproteins (ox-LDL), and biomechanical stimuli (low shear stress) lead to endothelial cell activation and dysfunction. Consequently, it is crucial to investigate how endothelial cells respond to different stressors and ways to alter endothelial cell activation in AS development, as they are the earliest cells to respond. Caveolin-1 (Cav1) is a 21-24-kDa membrane protein located in caveolae and highly expressed in endothelial cells, which plays a vital role in regulating lipid transport, inflammatory responses, and various cellular signaling pathways and has atherogenic effects. This review summarizes recent studies on the structure and physiological functions of Cav1 and outlines the potential mechanisms it mediates in AS development. Included are the roles of Cav1 in the regulation of endothelial cell autophagy, response to shear stress, modulation of the eNOS/NO axis, and transduction of inflammatory signaling pathways. This review provides a rationale for proposing Cav1 as a novel target for the prevention of AS, as well as new ideas for therapeutic strategies for early AS.
Keywords: Atherosclerosis; Autophagy; Caveolin-1; Endothelial cells; Inflammation; eNOS/NO axis.
© 2023 The Authors.
Conflict of interest statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures

References
-
- Tsao C.W., Aday A.W., Almarzooq Z.I., Alonso A., Beaton A.Z., Bittencourt M.S., Boehme A.K., Buxton A.E., Carson A.P., Commodore-Mensah Y., Elkind M.S.V., Evenson K.R., Eze-Nliam C., Ferguson J.F., Generoso G., Ho J.E., Kalani R., Khan S.S., Kissela B.M., Knutson K.L., Levine D.A., Lewis T.T., Liu J., Loop M.S., Ma J., Mussolino M.E., Navaneethan S.D., Perak A.M., Poudel R., Rezk-Hanna M., Roth G.A., Schroeder E.B., Shah S.H., Thacker E.L., VanWagner L.B., Virani S.S., Voecks J.H., Wang N.Y., Yaffe K., Martin S.S. Heart disease and stroke statistics-2022 update: a report from the American heart association. Circulation. 2022;145(8):e153–e639. doi: 10.1161/cir.0000000000001052. - DOI - PubMed
-
- Arora S., Stouffer G.A., Kucharska-Newton A.M., Qamar A., Vaduganathan M., Pandey A., Porterfield D., Blankstein R., Rosamond W.D., Bhatt D.L., Caughey M.C. Twenty year trends and sex differences in young adults hospitalized with acute myocardial infarction. Circulation. 2019;139(8):1047–1056. doi: 10.1161/circulationaha.118.037137. - DOI - PMC - PubMed
-
- Collaborators GMaCoD Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet (London, England) 2015;385(9963):117–171. doi: 10.1016/s0140-6736(14)61682-2. - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
