

Identification of the novel FOXP3-dependent Treg cell transcription factor MEOX1 by high-dimensional analysis of human CD4+ T cells
- PMID: 37559728
- PMCID: PMC10407399
- DOI: 10.3389/fimmu.2023.1107397
Identification of the novel FOXP3-dependent Treg cell transcription factor MEOX1 by high-dimensional analysis of human CD4+ T cells
Abstract
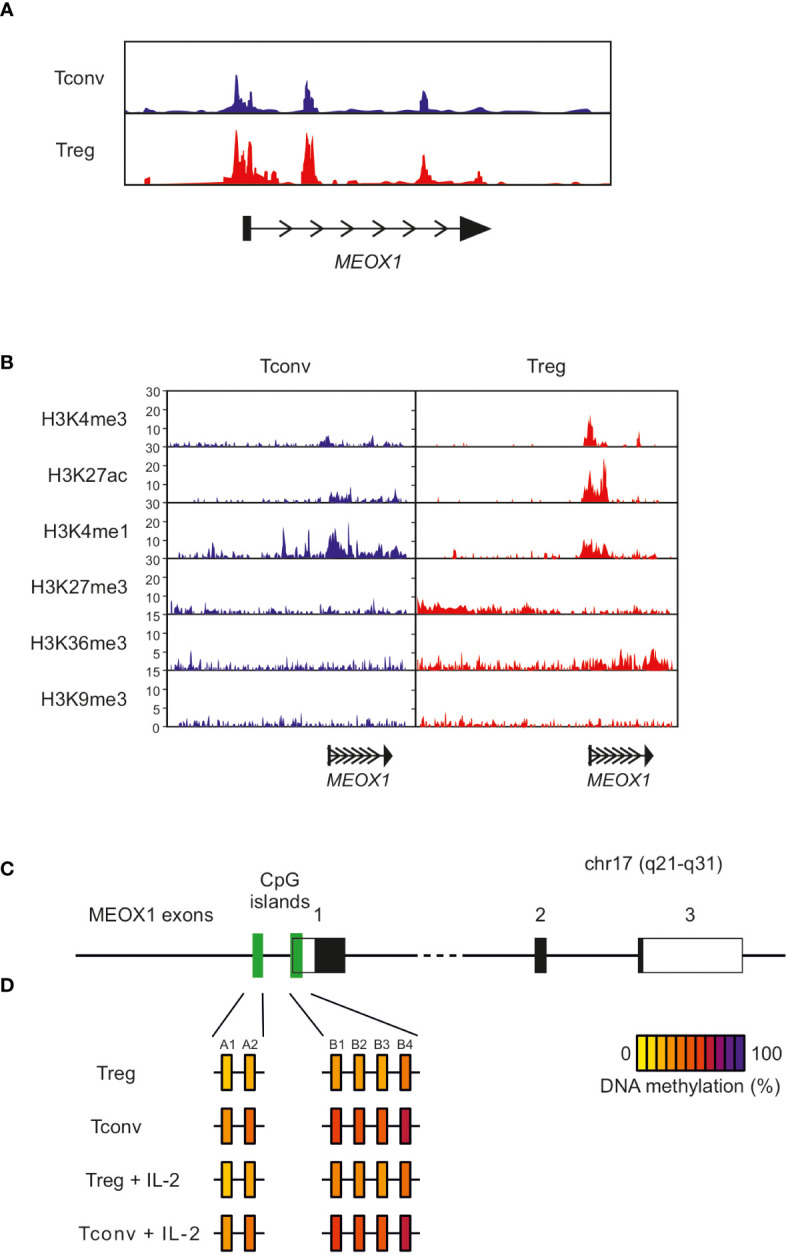
CD4+ T cells play a central role in the adaptive immune response through their capacity to activate, support and control other immune cells. Although these cells have become the focus of intense research, a comprehensive understanding of the underlying regulatory networks that orchestrate CD4+ T cell function and activation is still incomplete. Here, we analyzed a large transcriptomic dataset consisting of 48 different human CD4+ T cell conditions. By performing reverse network engineering, we identified six common denominators of CD4+ T cell functionality (CREB1, E2F3, AHR, STAT1, NFAT5 and NFATC3). Moreover, we also analyzed condition-specific genes which led us to the identification of the transcription factor MEOX1 in Treg cells. Expression of MEOX1 was comparable to FOXP3 in Treg cells and can be upregulated by IL-2. Epigenetic analyses revealed a permissive epigenetic landscape for MEOX1 solely in Treg cells. Knockdown of MEOX1 in Treg cells revealed a profound impact on downstream gene expression programs and Treg cell suppressive capacity. These findings in the context of CD4+ T cells contribute to a better understanding of the transcriptional networks and biological mechanisms controlling CD4+ T cell functionality, which opens new avenues for future therapeutic strategies.
Keywords: Foxp3; MEOX1; Treg cells; human CD4; regulatory T cells.
Copyright © 2023 Baßler, Schmidleithner, Shakiba, Elmzzahi, Köhne, Floess, Scholz, Ohkura, Sadlon, Klee, Neubauer, Sakaguchi, Barry, Huehn, Bonaguro, Ulas and Beyer.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures






References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous