

DNA methylation networks underlying mammalian traits
- PMID: 37561875
- PMCID: PMC11180965
- DOI: 10.1126/science.abq5693
DNA methylation networks underlying mammalian traits
Abstract
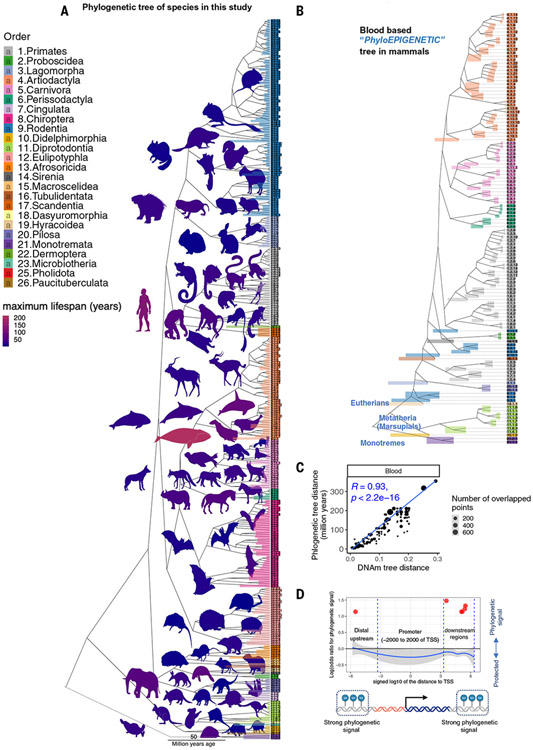
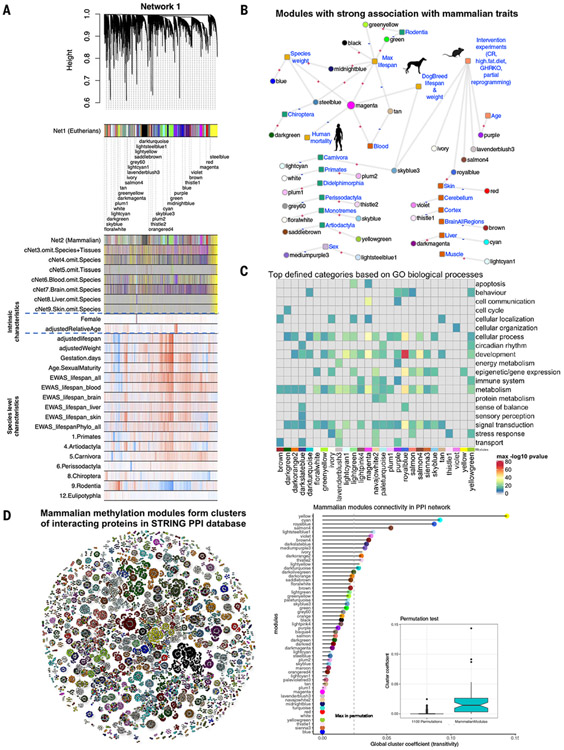
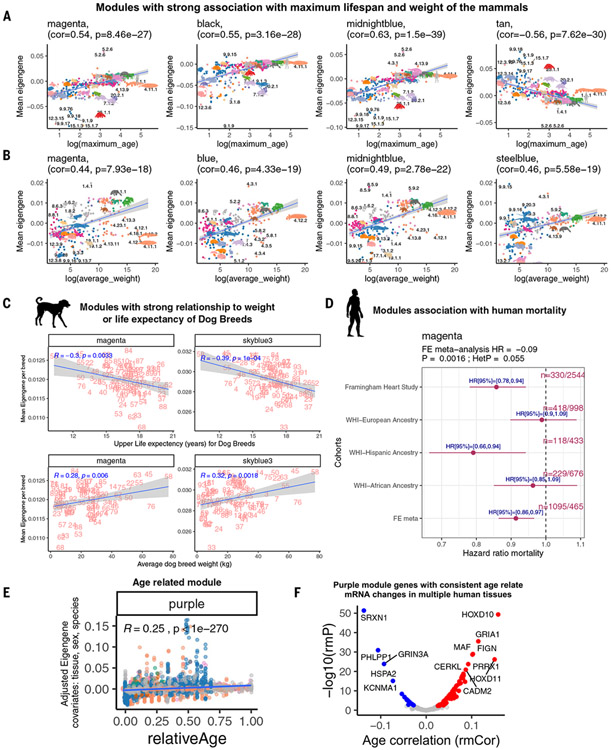
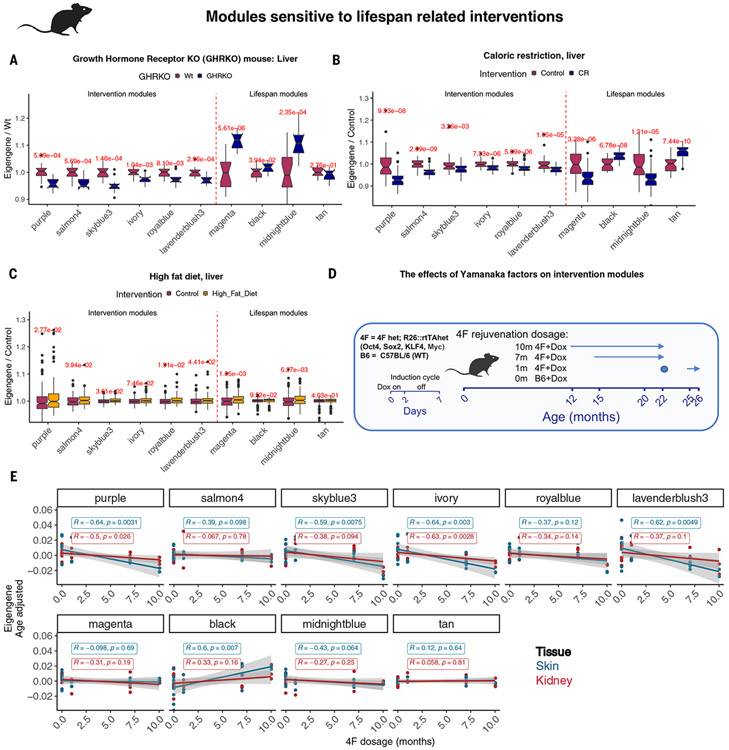
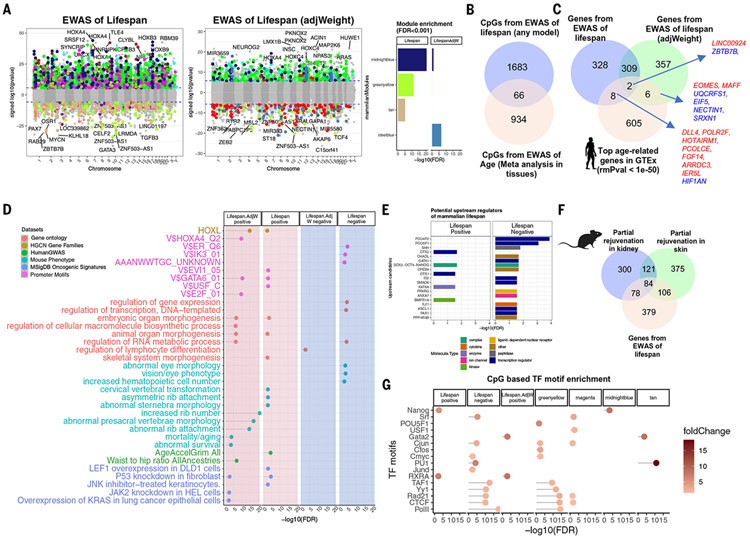
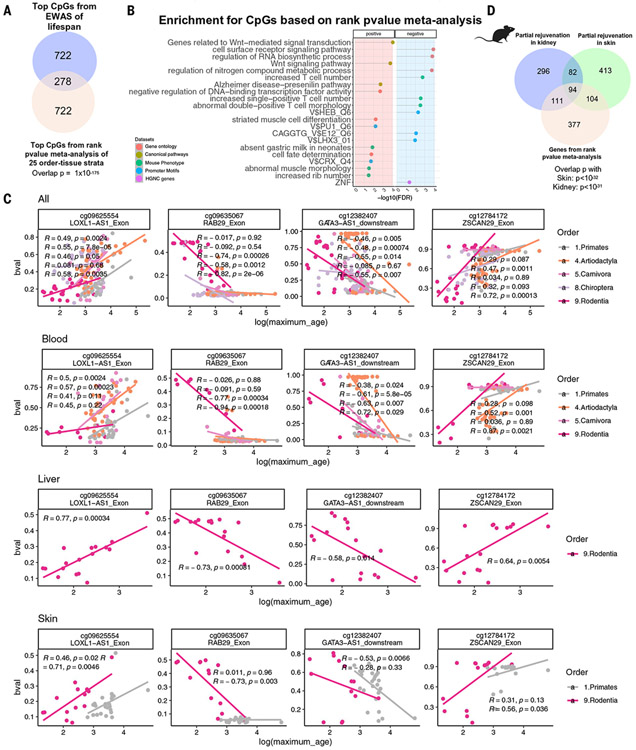
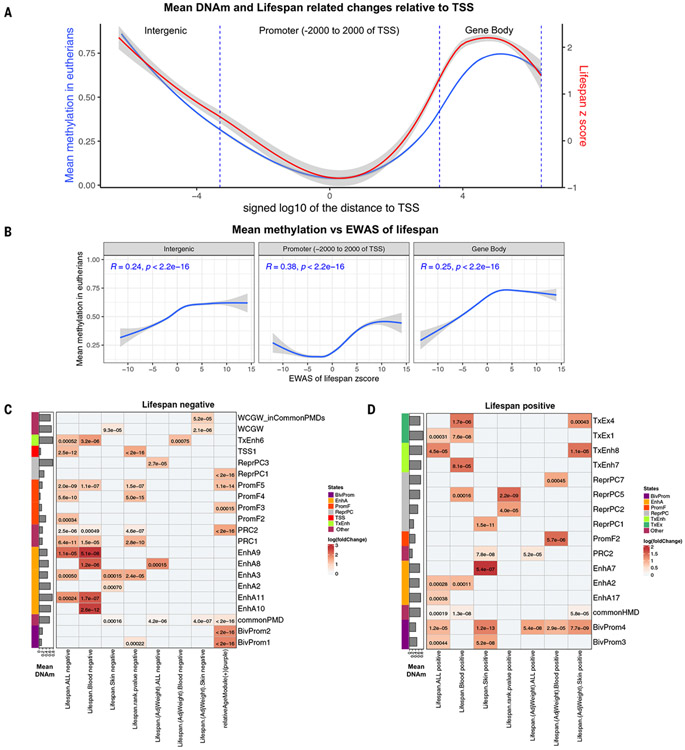
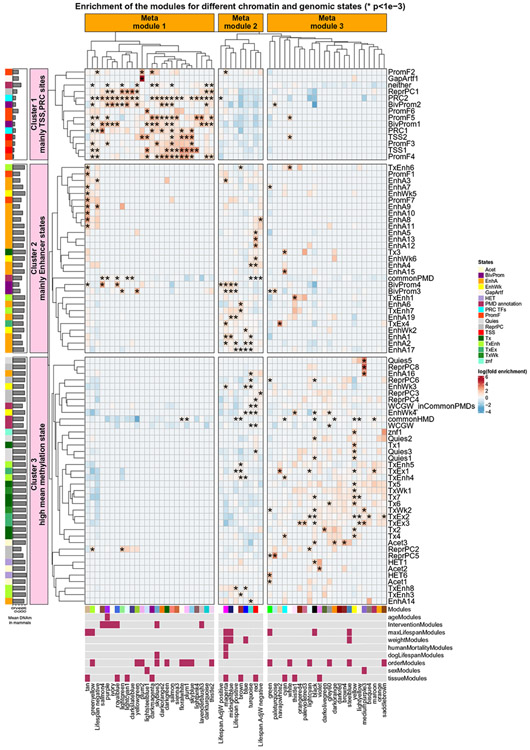
Using DNA methylation profiles (n = 15,456) from 348 mammalian species, we constructed phyloepigenetic trees that bear marked similarities to traditional phylogenetic ones. Using unsupervised clustering across all samples, we identified 55 distinct cytosine modules, of which 30 are related to traits such as maximum life span, adult weight, age, sex, and human mortality risk. Maximum life span is associated with methylation levels in HOXL subclass homeobox genes and developmental processes and is potentially regulated by pluripotency transcription factors. The methylation state of some modules responds to perturbations such as caloric restriction, ablation of growth hormone receptors, consumption of high-fat diets, and expression of Yamanaka factors. This study reveals an intertwined evolution of the genome and epigenome that mediates the biological characteristics and traits of different mammalian species.
Figures
References
MeSH terms
Grants and funding
- R37 AG046320/AG/NIA NIH HHS/United States
- R01 AG043930/AG/NIA NIH HHS/United States
- R21 AG055841/AG/NIA NIH HHS/United States
- R56 AG072736/AG/NIA NIH HHS/United States
- R01 AG027237/AG/NIA NIH HHS/United States
- P30 AG044271/AG/NIA NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
- R21 ES024356/ES/NIEHS NIH HHS/United States
- U01 AG060908/AG/NIA NIH HHS/United States
- R01 AG045795/AG/NIA NIH HHS/United States
- U19 AG057758/AG/NIA NIH HHS/United States
- R01 AG065403/AG/NIA NIH HHS/United States
- P01 AG047200/AG/NIA NIH HHS/United States
- U34 AG068482/AG/NIA NIH HHS/United States
- R21 DE028271/DE/NIDCR NIH HHS/United States
- R01 AG067782/AG/NIA NIH HHS/United States
- P30 AG013319/AG/NIA NIH HHS/United States
- R01 AG050797/AG/NIA NIH HHS/United States
- R21 AG078784/AG/NIA NIH HHS/United States
- R21 OD022988/OD/NIH HHS/United States
- R01 AG072736/AG/NIA NIH HHS/United States
- 20412/CRUK_/Cancer Research UK/United Kingdom
- R56 AG076607/AG/NIA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
