

Copy number architectures define treatment-mediated selection of lethal prostate cancer clones
- PMID: 37563129
- PMCID: PMC10415299
- DOI: 10.1038/s41467-023-40315-9
Copy number architectures define treatment-mediated selection of lethal prostate cancer clones
Abstract
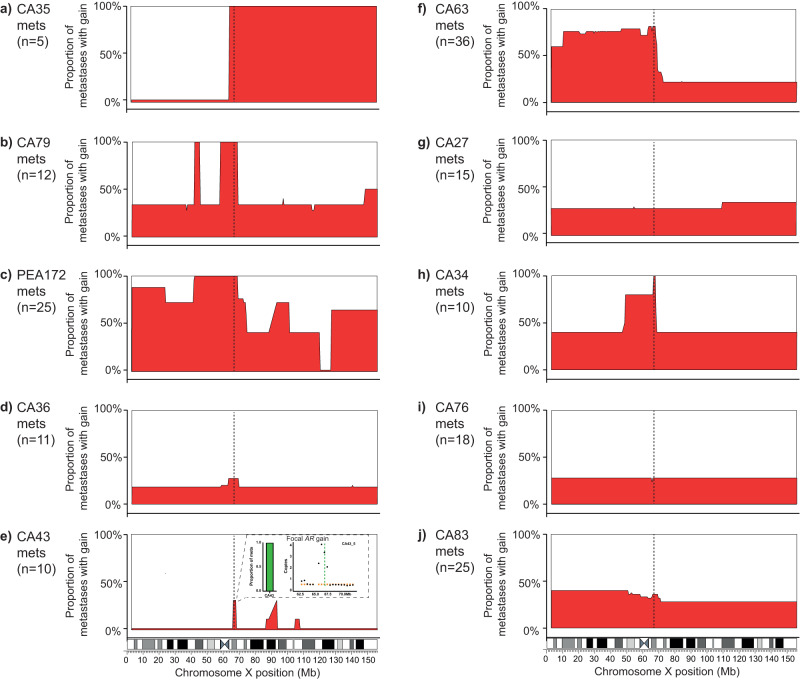
Despite initial responses to hormone treatment, metastatic prostate cancer invariably evolves to a lethal state. To characterize the intra-patient evolutionary relationships of metastases that evade treatment, we perform genome-wide copy number profiling and bespoke approaches targeting the androgen receptor (AR) on 167 metastatic regions from 11 organs harvested post-mortem from 10 men who died from prostate cancer. We identify diverse and patient-unique alterations clustering around the AR in metastases from every patient with evidence of independent acquisition of related genomic changes within an individual and, in some patients, the co-existence of AR-neutral clones. Using the genomic boundaries of pan-autosome copy number changes, we confirm a common clone of origin across metastases and diagnostic biopsies, and identified in individual patients, clusters of metastases occupied by dominant clones with diverged autosomal copy number alterations. These autosome-defined clusters are characterized by cluster-specific AR gene architectures, and in two index cases are topologically more congruent than by chance (p-values 3.07 × 10-8 and 6.4 × 10-4). Integration with anatomical sites suggests patterns of spread and points of genomic divergence. Here, we show that copy number boundaries identify treatment-selected clones with putatively distinct lethal trajectories.
© 2023. Springer Nature Limited.
Conflict of interest statement
M.J.-H. has received funding from CRUK, NIH National Cancer Institute, IASLC International Lung Cancer Foundation, Lung Cancer Research Foundation, Rosetrees Trust, UKI NETs and NIHR. M.J.-H. has consulted for, and is a member of, the Achilles Therapeutics Scientific Advisory Board and Steering Committee, has received speaker honoraria from Pfizer, Astex Pharmaceuticals, Oslo Cancer Cluster, Bristol Myers Squibb, and is co-inventor on a European patent application relating to methods to detect lung cancer (PCT/US2017/028013). C.S. acknowledges grants from AstraZeneca, Boehringer-Ingelheim, Bristol Myers Squibb, Pfizer, Roche-Ventana, Invitae (previously Archer Dx Inc—collaboration in minimal residual disease sequencing technologies), Ono Pharmaceutical, and Personalis. He is Chief Investigator for the AZ MeRmaiD 1 and 2 clinical trials and is the Steering Committee Chair. He is also Co-Chief Investigator of the NHS Galleri trial funded by GRAIL and a paid member of GRAIL’s Scientific Advisory Board. He receives consultant fees from Achilles Therapeutics (also SAB member), Bicycle Therapeutics (also a SAB member), Genentech, Medicxi, China Innovation Centre of Roche (CICoR) formerly Roche Innovation Centre—Shanghai, Metabomed (until July 2022), and the Sarah Cannon Research Institute C.S has received honoraria from Amgen, AstraZeneca, Bristol Myers Squibb, GlaxoSmithKline, Illumina, MSD, Novartis, Pfizer, and Roche-Ventana. C.S. has previously held stock options in Apogen Biotechnologies and GRAIL, and currently has stock options in Epic Bioscience, Bicycle Therapeutics, and has stock options and is co-founder of Achilles Therapeutics. C.S. also declares a patent application (PCT/US2017/028013) for methods to lung cancer; targeting neoantigens (PCT/EP2016/059401); identifying patent response to immune checkpoint blockade (PCT/EP2016/071471), determining HLA LOH (PCT/GB2018/052004); predicting survival rates of patients with cancer (PCT/GB2020/050221), identifying patients who respond to cancer treatment (PCT/GB2018/051912); methods for lung cancer detection (US20190106751A1). C.S. is an inventor on a European patent application (PCT/GB2017/053289) relating to assay technology to detect tumor recurrence. This patent has been licensed to a commercial entity and under their terms of employment C.S is due a revenue share of any revenue generated from such license(s). M.L. reports grants or contracts from BMS, Shionogi, and AstraZeneca; consulting fees from BioNTech, Bicycle Therapeutics, Janssen, Merck Sorano, Pfizer, and ADC Therapeutics; honoraria from AstraZeneca and Pfizer; and support for attending meetings or travel from MSD, Janssen, and Bayer. S.S. has served on advisory boards for Bristol Myer Squibb, Merck Sharp and Dohme, Astra Zeneca, Janssen and has received institutional grant funding from Merck Sharp and Dohme, Astra Zeneca, Amgen, and Advanced Accelerator Applications (AAA), a Novartis Company, Merck Serono and Roche/Genentech (outside the submitted work). P.C., D.W., and G.A. have a patent on blood methylation markers (GB1915469.9). G.A. received personal fees, grants, and travel support from Janssen and Astellas Pharma; personal fees or travel support from Pfizer, Novartis/AAA, Bayer Healthcare Pharmaceuticals, AstraZeneca, and Sanofi-Aventis; in addition, G.A.’s former employer, The Institute of Cancer Research, receives royalty income from abiraterone and G.A. receives a share of this income through the Institute’s Rewards to Discoverers Scheme. G.A. has received research funding (institutional) from Janssen, Astellas Pharma, and Novartis. All other authors declare no potential conflicts of interest.
Figures




References
-
- Visakorpi T, et al. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat. Genet. 1995;9:401–406. - PubMed
-
- Annala M, et al. Evolution of castration-resistant prostate cancer in ctDNA during sequential androgen receptor pathway inhibition. Clin. Cancer Res. 2021;27:4610–4623. - PubMed
-
- Taplin ME, et al. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N. Engl. J. Med. 1995;332:1393–1398. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
