

Universal DNA methylation age across mammalian tissues
- PMID: 37563227
- PMCID: PMC10501909
- DOI: 10.1038/s43587-023-00462-6
Universal DNA methylation age across mammalian tissues
Erratum in
-
Author Correction: Universal DNA methylation age across mammalian tissues.Nat Aging. 2023 Nov;3(11):1462. doi: 10.1038/s43587-023-00499-7. Nat Aging. 2023. PMID: 37674040 Free PMC article. No abstract available.
Abstract
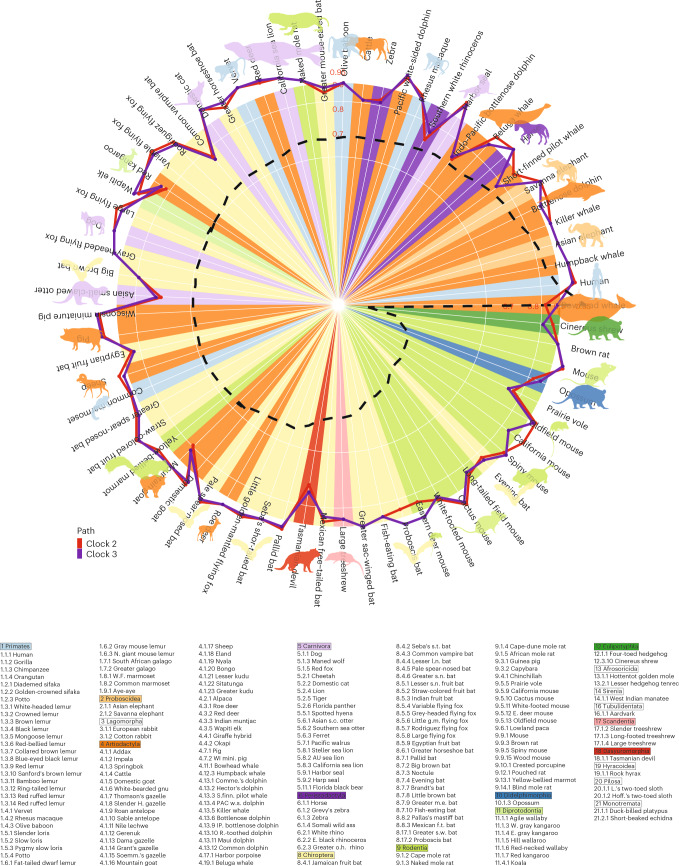
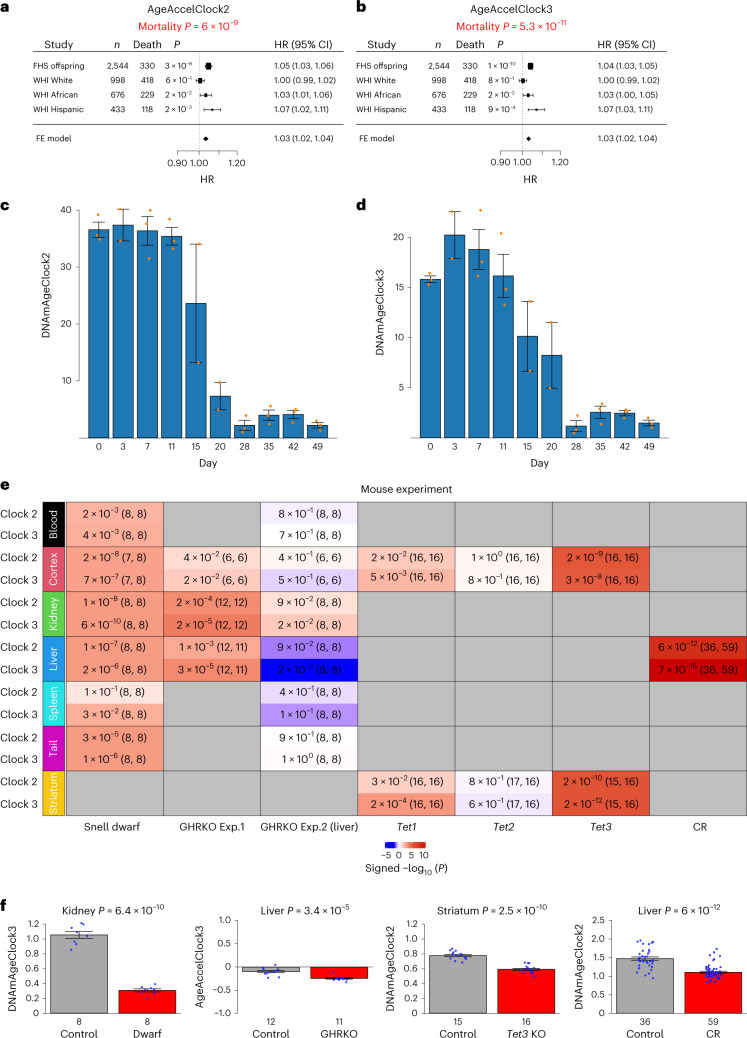
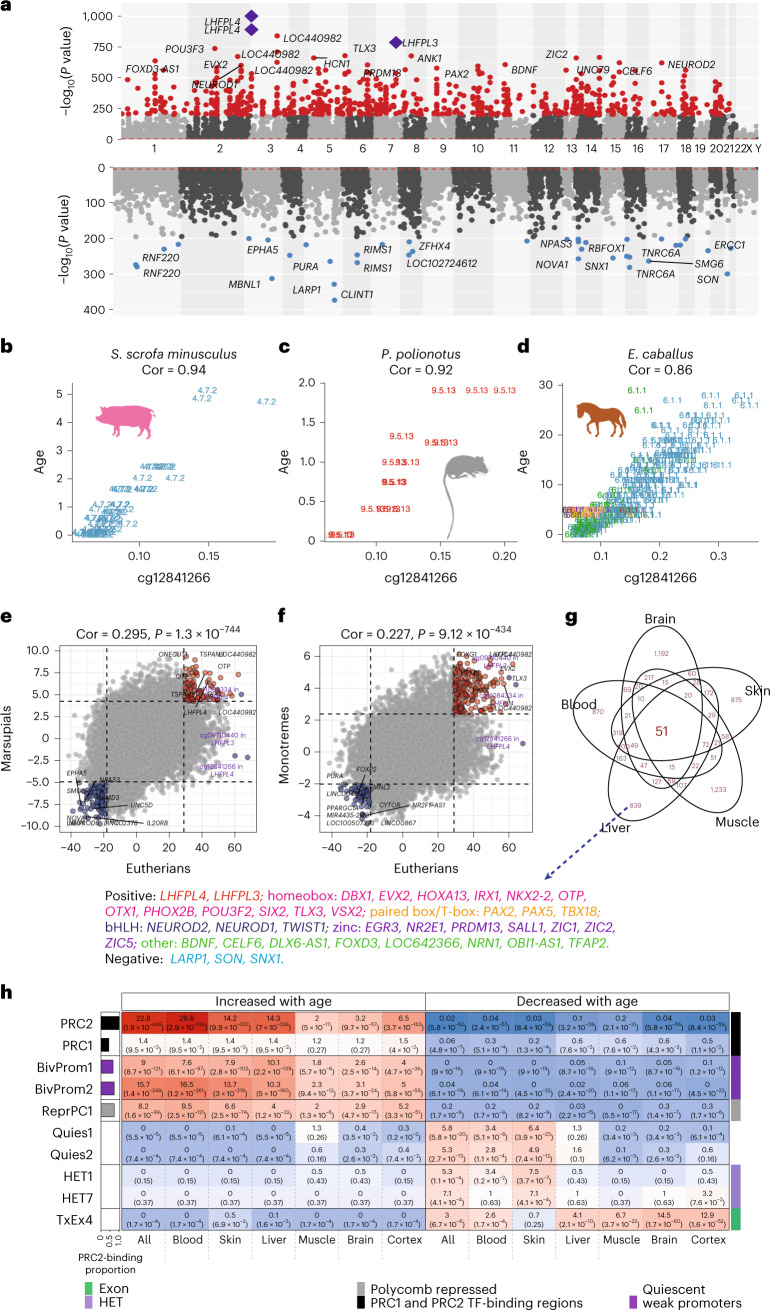
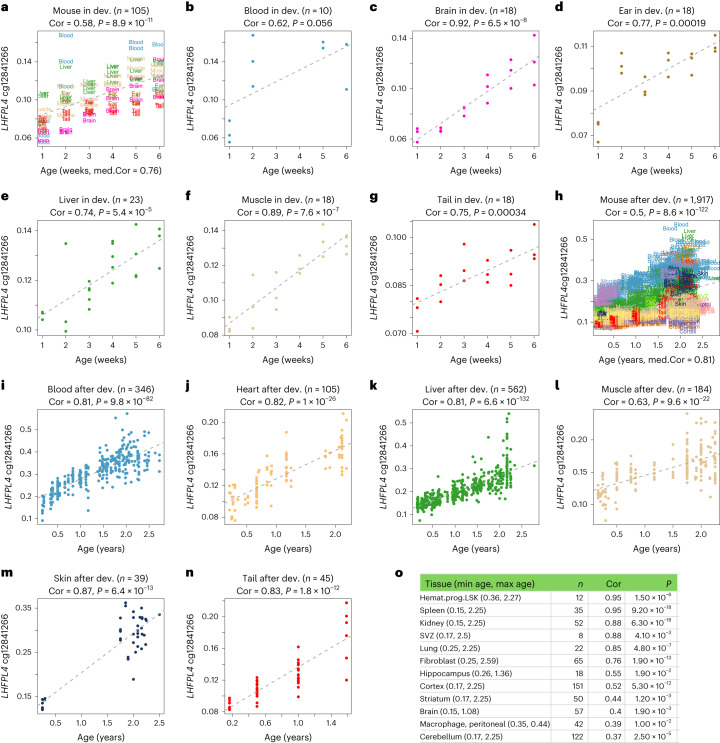
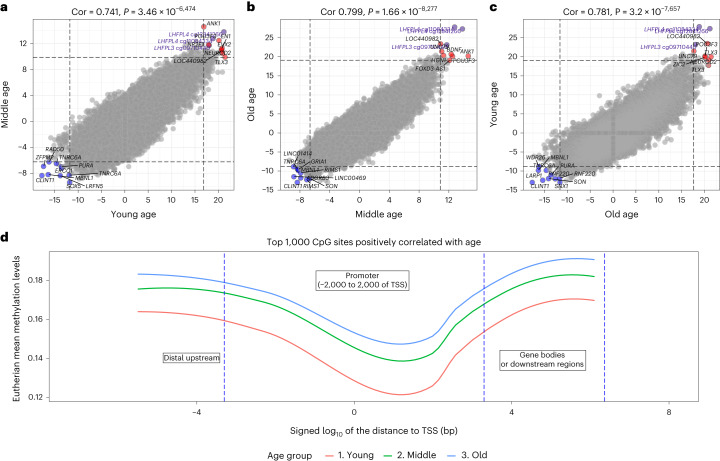
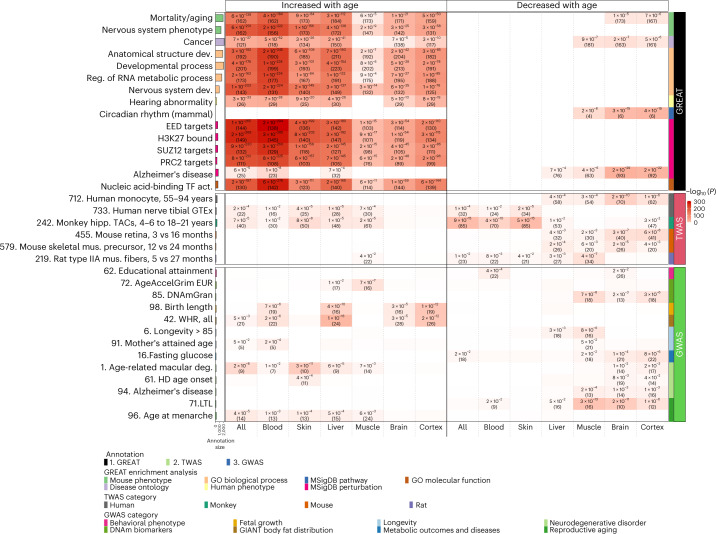
Aging, often considered a result of random cellular damage, can be accurately estimated using DNA methylation profiles, the foundation of pan-tissue epigenetic clocks. Here, we demonstrate the development of universal pan-mammalian clocks, using 11,754 methylation arrays from our Mammalian Methylation Consortium, which encompass 59 tissue types across 185 mammalian species. These predictive models estimate mammalian tissue age with high accuracy (r > 0.96). Age deviations correlate with human mortality risk, mouse somatotropic axis mutations and caloric restriction. We identified specific cytosines with methylation levels that change with age across numerous species. These sites, highly enriched in polycomb repressive complex 2-binding locations, are near genes implicated in mammalian development, cancer, obesity and longevity. Our findings offer new evidence suggesting that aging is evolutionarily conserved and intertwined with developmental processes across all mammals.
© 2023. The Author(s).
Conflict of interest statement
The Regents of the University of California filed a patent application (publication number WO2020150705) related to this work on which S.H., A. Arneson and J.E. are named inventors. S.H. and R.T.B. are founders of the non-profit Epigenetic Clock Development Foundation, which has licensed several patents from UC Regents, and distributes the mammalian methylation array. The remaining authors declare no competing interests.
Figures












References
Publication types
MeSH terms
Grants and funding
- MR/T021985/1/MRC_/Medical Research Council/United Kingdom
- R21 AG055841/AG/NIA NIH HHS/United States
- P30 AG013319/AG/NIA NIH HHS/United States
- N01 HC025195/HL/NHLBI NIH HHS/United States
- HHSN268201600003C/HL/NHLBI NIH HHS/United States
- HHSN268201500001I/HL/NHLBI NIH HHS/United States
- HHSN268201600004C/HL/NHLBI NIH HHS/United States
- P01 AG051449/AG/NIA NIH HHS/United States
- P51 OD011133/OD/NIH HHS/United States
- K00 CA234840/CA/NCI NIH HHS/United States
- R37 AG046320/AG/NIA NIH HHS/United States
- U01 AG060908/AG/NIA NIH HHS/United States
- HHSN268201600002C/HL/NHLBI NIH HHS/United States
- U19 AG057758/AG/NIA NIH HHS/United States
- R01 AG065403/AG/NIA NIH HHS/United States
- HHSN268201500001C/HL/NHLBI NIH HHS/United States
- HHSN268201600018C/HL/NHLBI NIH HHS/United States
- P01 AG047200/AG/NIA NIH HHS/United States
- U34 AG068482/AG/NIA NIH HHS/United States
- R01 AG050797/AG/NIA NIH HHS/United States
- R21 AG078784/AG/NIA NIH HHS/United States
- P30 AG044271/AG/NIA NIH HHS/United States
- HHSN268201600001C/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
